This tutorial will serve as a guideline for how to go about analyzing RNA sequencing data when a reference genome is available. We will be going through quality control of the reads, alignment of the reads to the reference genome, conversion of the files to raw counts, analysis of the counts with DeSeq2 and DEXSeq, and finally annotation of the reads using Biomart. Most of this will be done on the BBC server unless otherwise stated.
The packages we’ll be using can be found here: Page by Dister Deoss
The data we will be using are comparative transcriptomes of soybeans grown at either ambient or elevated O3 levels. Each condition was done in triplicate, giving us a total of six samples we will be working with. The paper that these samples come from (which also serves as a great background reading on RNA-seq) can be found here:
The Bench Scientist’s Guide to statistical Analysis of RNA-Seq Data
The samples we will be using are described by the following accession numbers; SRR391535, SRR391536, SRR391537, SRR391538, SRR391539, and SRR391541. They can be found in results 13 through 18 of the following NCBI search:
http://www.ncbi.nlm.nih.gov/sra/?term=SRP009826
The script for downloading these .SRA files and converting them to fastq can be found in
/common/RNASeq_Workshop/Soybean/Quality_Control as the file fastq-dump.sh. The fastq files themselves are also already saved to this same directory.
module load sratoolkit/2.8.1 fastq-dump SRR391535
Repeat fastq-dump for SRR391536, SRR391537, SRR391538, SRR391539, and SRR391541
Quality Control on the Reads Using Sickle:
Step one is to perform quality control on the reads using Sickle. We are using unpaired reads, as indicated by the “se” flag in the script below. The -f flag designates the input file, -o is the output file, -q is our minimum quality score and -l is the minimum read length. The trimmed output files are what we will be using for the next steps of our analysis.
sickle se -f SRR391535.fastq -t sanger -o trimmed_SRR391535.fastq -q 35 -l 45
Repeat this command for SRR391536, SRR391537, SRR391538, SRR391539, and SRR39154. The script for running quality control on all six of our samples can be found in
/common/RNASeq_Workshop/Soybean/Quality_Control as the file sickle_soybean.sh. The output trimmed fastq files are also stored in this directory.
Alignment of Trimmed Reads Using STAR:
For this next step, you will first need to download the reference genome and annotation file for Glycine max (soybean). The files I used can be found at the following link:
You will need to create a user name and password for this database before you download the files. Once you’ve done that, you can download the assembly file Gmax_275_v2, and the annotation file Gmax_275_Wm82.a2.v1.gene_exons. Having the correct files is important for annotating the genes with Biomart later on.
Now that you have the genome and annotation files, you will create a genome index using the following script:
STAR --runMode genomeGenerate --genomeDir /common/RNASeq_Workshop/Soybean/gmax_genome/ --genomeFastaFiles /common/RNASeq_Workshop/Soybean/gmax_genome/Gmax_275_v2 --sjdbGTFfile /common/RNASeq_Workshop/Soybean/gmax_genome/Gmax_275_Wm82.a2.v1.gene_exons --sjdbGTFtagExonParentTranscript Parent --sjdbOverhang 100 --runThreadN 8
You will likely have to alter this script slightly to reflect the directory that you are working in and the specific names you gave your files, but the general idea is there. Indexing the genome allows for more efficient mapping of the reads to the genome.
The assembly file, annotation file, as well as all of the files created from indexing the genome can be found in
/common/RNASeq_Workshop/Soybean/gmax_genome
Now that you have your genome indexed, you can begin mapping your trimmed reads with the following script:
STAR --genomeDir /common/RNASeq_Workshop/Soybean/gmax_genome/ --readFilesIn trimmed_SRR391535.fastq --runThreadN 8 --outSAMtype BAM SortedByCoordinate --outFileNamePrefix SRR391535
The –genomeDir flag refers to the directory in which your indexed genome is located. The output we get from this are .BAM files; binary files that will be converted to raw counts in our next step. Repeat it for SRR391536, SRR391537, SRR391538, SRR391539, and SRR391541.
The script for mapping all six of our trimmed reads to .bam files can be found in
/common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping as the file star_soybean.sh. The .bam output files are also stored in this directory.
Convert BAM Files to Raw Counts with HTSeq: We will use HTSeq to transform these mapped reads into counts that we can analyze with R. “-s” indicates we do not have strand specific counts. “-r” indicates the order that the reads were generated, for us it was by alignment position. “-t” indicates the feature from the annotation file we will be using, which in our case will be exons. “-i” indicates what attribute we will be using from the annotation file, here it is the PAC transcript ID. We identify that we are pulling in a .bam file (“-f bam”) and proceed to identify, and say where it will go.
htseq-count -s no -r pos —t exon -i pacid -f bam SRR391535Aligned.sortedByCoord.out.bam /common/RNASeq_Workshop/Soybean/gmax_genome/Gmax_275_Wm82.a2.v1.gene_exons > SRR391535-output_basename.counts
The script to generate all 6 .count files is located in
/common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping as the file htseq_soybean.sh. The .count output files are saved in
/common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping/counts
Analysis of Counts with DESeq2:
For the remaining steps I find it easier to to work from a desktop rather than the server. So you can download the .count files you just created from the server onto your computer. You will also need to download R to run DESeq2, and I’d also recommend installing RStudio, which provides a graphical interface that makes working with R scripts much easier. They can be found here:
The R DESeq2 library also must be installed. To install this package, start the R console and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("DESeq2")
The R code below is long and slightly complicated, but I will highlight major points. This script was adapted from here and here, and much credit goes to those authors. Some important notes:
- The most important information comes out as “-replaceoutliers-results.csv” there we can see adjusted and normal p-values, as well as log2foldchange for all of the genes.
- par(mar) manipulation is used to make the most appealing figures, but these values are not the same for every display or system or figure. Much documentation is available online on how to manipulate and best use par() and ggplot2 graphing parameters: DESeq2 Manual
# Load DESeq2 library library("DESeq2")
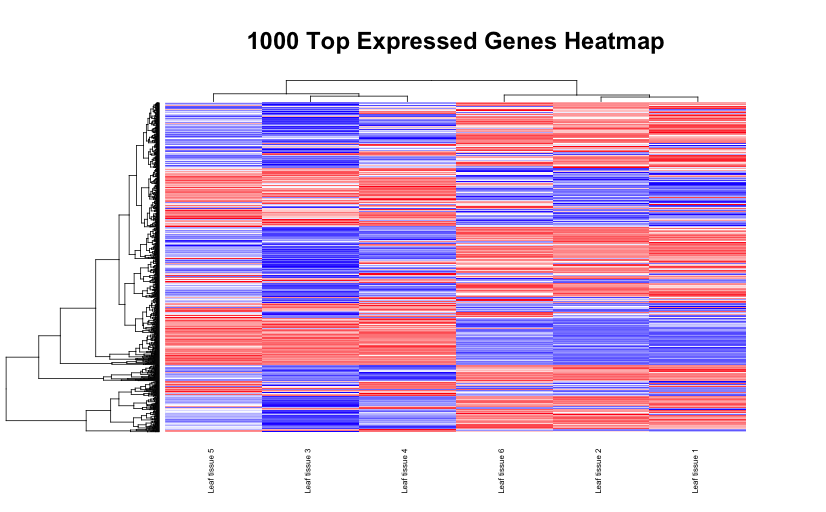
# Set the working directory directory <- "~/Documents/counts2" setwd(directory) # Set the prefix for each output file name outputPrefix <- "Gmax_DESeq2" sampleFiles<- c("SRR391535-output_basename.counts", "SRR391536-output_basename.counts", "SRR391537-output_basename.counts", "SRR391538-output_basename.counts", "SRR391539-output_basename.counts", "SRR391541-output_basename.counts") sampleNames <- c("Leaf tissue 1","Leaf tissue 2","Leaf tissue 3","Leaf tissue 4","Leaf tissue 5","Leaf tissue 6") sampleCondition <- c("ambient","ambient","elevated","elevated","elevated","ambient") sampleTable <- data.frame(sampleName = sampleNames, fileName = sampleFiles, condition = sampleCondition) treatments = c("ambient","elevated") library("DESeq2") ddsHTSeq <- DESeqDataSetFromHTSeqCount(sampleTable = sampleTable, directory = directory, design = ~ condition) colData(ddsHTSeq)$condition <- factor(colData(ddsHTSeq)$condition, levels = treatments) #guts dds <- DESeq(ddsHTSeq) res <- results(dds) # copied from: https://benchtobioinformatics.wordpress.com/category/dexseq/ # order results by padj value (most significant to least) res= subset(res, padj<0.05) res <- res[order(res$padj),] # should see DataFrame of baseMean, log2Foldchange, stat, pval, padj # save data results and normalized reads to csv resdata <- merge(as.data.frame(res), as.data.frame(counts(dds,normalized =TRUE)), by = 'row.names', sort = FALSE) names(resdata)[1] <- 'gene' write.csv(resdata, file = paste0(outputPrefix, "-results-with-normalized.csv")) # send normalized counts to tab delimited file for GSEA, etc. write.table(as.data.frame(counts(dds),normalized=T), file = paste0(outputPrefix, "_normalized_counts.txt"), sep = '\t') # produce DataFrame of results of statistical tests mcols(res, use.names = T) write.csv(as.data.frame(mcols(res, use.name = T)),file = paste0(outputPrefix, "-test-conditions.csv")) # replacing outlier value with estimated value as predicted by distrubution using # "trimmed mean" approach. recommended if you have several replicates per treatment # DESeq2 will automatically do this if you have 7 or more replicates ddsClean <- replaceOutliersWithTrimmedMean(dds) ddsClean <- DESeq(ddsClean) tab <- table(initial = results(dds)$padj < 0.05, cleaned = results(ddsClean)$padj < 0.05) addmargins(tab) write.csv(as.data.frame(tab),file = paste0(outputPrefix, "-replaceoutliers.csv")) resClean <- results(ddsClean) resClean = subset(res, padj<0.05) resClean <- resClean[order(resClean$padj),] write.csv(as.data.frame(resClean),file = paste0(outputPrefix, "-replaceoutliers-results.csv")) #################################################################################### # Exploratory data analysis of RNAseq data with DESeq2 # # these next R scripts are for a variety of visualization, QC and other plots to # get a sense of what the RNAseq data looks like based on DESEq2 analysis # # 1) MA plot # 2) rlog stabilization and variance stabiliazation # 3) variance stabilization plot # 4) heatmap of clustering analysis # 5) PCA plot # # #################################################################################### # MA plot of RNAseq data for entire dataset # http://en.wikipedia.org/wiki/MA_plot # genes with padj < 0.1 are colored Red plotMA(dds, ylim=c(-8,8),main = "RNAseq experiment") dev.copy(png, paste0(outputPrefix, "-MAplot_initial_analysis.png")) dev.off() # transform raw counts into normalized values # DESeq2 has two options: 1) rlog transformed and 2) variance stabilization # variance stabilization is very good for heatmaps, etc. rld <- rlogTransformation(dds, blind=T) vsd <- varianceStabilizingTransformation(dds, blind=T) # save normalized values write.table(as.data.frame(assay(rld),file = paste0(outputPrefix, "-rlog-transformed-counts.txt"), sep = '\t')) write.table(as.data.frame(assay(vsd),file = paste0(outputPrefix, "-vst-transformed-counts.txt"), sep = '\t')) # plot to show effect of transformation # axis is square root of variance over the mean for all samples par(mai = ifelse(1:4 <= 2, par('mai'),0)) px <- counts(dds)[,1] / sizeFactors(dds)[1] ord <- order(px) ord <- ord[px[ord] < 150] ord <- ord[seq(1,length(ord),length=50)] last <- ord[length(ord)] vstcol <- c('blue','black') matplot(px[ord], cbind(assay(vsd)[,1], log2(px))[ord, ],type='l', lty = 1, col=vstcol, xlab = 'n', ylab = 'f(n)') legend('bottomright',legend=c(expression('variance stabilizing transformation'), expression(log[2](n/s[1]))), fill=vstcol) dev.copy(png,paste0(outputPrefix, "-variance_stabilizing.png")) dev.off() # clustering analysis # excerpts from http://dwheelerau.com/2014/02/17/how-to-use-deseq2-to-analyse-rnaseq-data/ library("RColorBrewer") library("gplots") distsRL <- dist(t(assay(rld))) mat <- as.matrix(distsRL) rownames(mat) <- colnames(mat) <- with(colData(dds), paste(condition, sampleNames, sep=" : ")) #Or if you want conditions use: #rownames(mat) <- colnames(mat) <- with(colData(dds),condition) hmcol <- colorRampPalette(brewer.pal(9, "GnBu"))(100) dev.copy(png, paste0(outputPrefix, "-clustering.png")) heatmap.2(mat, trace = "none", col = rev(hmcol), margin = c(13,13)) dev.off() #Principal components plot shows additional but rough clustering of samples library("genefilter") library("ggplot2") library("grDevices") rv <- rowVars(assay(rld)) select <- order(rv, decreasing=T)[seq_len(min(500,length(rv)))] pc <- prcomp(t(assay(vsd)[select,])) # set condition condition <- treatments scores <- data.frame(pc$x, condition) (pcaplot <- ggplot(scores, aes(x = PC1, y = PC2, col = (factor(condition)))) + geom_point(size = 5) + ggtitle("Principal Components") + scale_colour_brewer(name = " ", palette = "Set1") + theme( plot.title = element_text(face = 'bold'), legend.position = c(.9,.2), legend.key = element_rect(fill = 'NA'), legend.text = element_text(size = 10, face = "bold"), axis.text.y = element_text(colour = "Black"), axis.text.x = element_text(colour = "Black"), axis.title.x = element_text(face = "bold"), axis.title.y = element_text(face = 'bold'), panel.grid.major.x = element_blank(), panel.grid.major.y = element_blank(), panel.grid.minor.x = element_blank(), panel.grid.minor.y = element_blank(), panel.background = element_rect(color = 'black',fill = NA) )) ggsave(pcaplot,file=paste0(outputPrefix, "-ggplot2.pdf")) # scatter plot of rlog transformations between Sample conditions # nice way to compare control and experimental samples head(assay(rld)) # plot(log2(1+counts(dds,normalized=T)[,1:2]),col='black',pch=20,cex=0.3, main='Log2 transformed') plot(assay(rld)[,1:3],col='#00000020',pch=20,cex=0.3, main = "rlog transformed") plot(assay(rld)[,2:4],col='#00000020',pch=20,cex=0.3, main = "rlog transformed") plot(assay(rld)[,6:5],col='#00000020',pch=20,cex=0.3, main = "rlog transformed") # heatmap of data library("RColorBrewer") library("gplots") # 1000 top expressed genes with heatmap.2 select <- order(rowMeans(counts(ddsClean,normalized=T)),decreasing=T)[1:1000] my_palette <- colorRampPalette(c("blue",'white','red'))(n=1000) heatmap.2(assay(vsd)[select,], col=my_palette, scale="row", key=T, keysize=1, symkey=T, density.info="none", trace="none", cexCol=0.6, labRow=F, main="1000 Top Expressed Genes Heatmap") dev.copy(png, paste0(outputPrefix, "-HEATMAP.png")) dev.off()
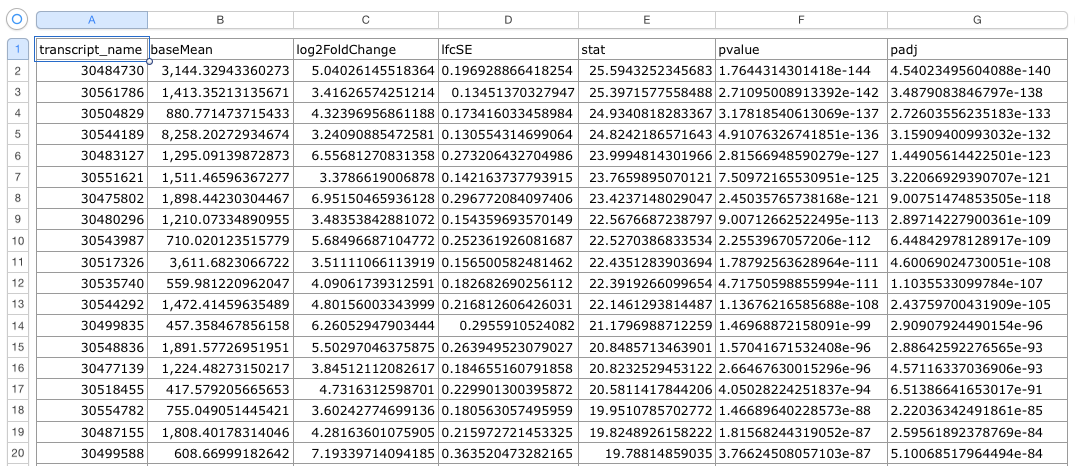
The .csv output file that you get from this R code should look something like this:
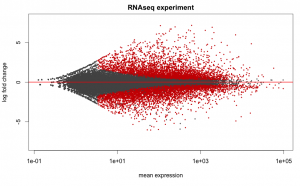
Below are some examples of the types of plots you can generate from RNAseq data using DESeq2:
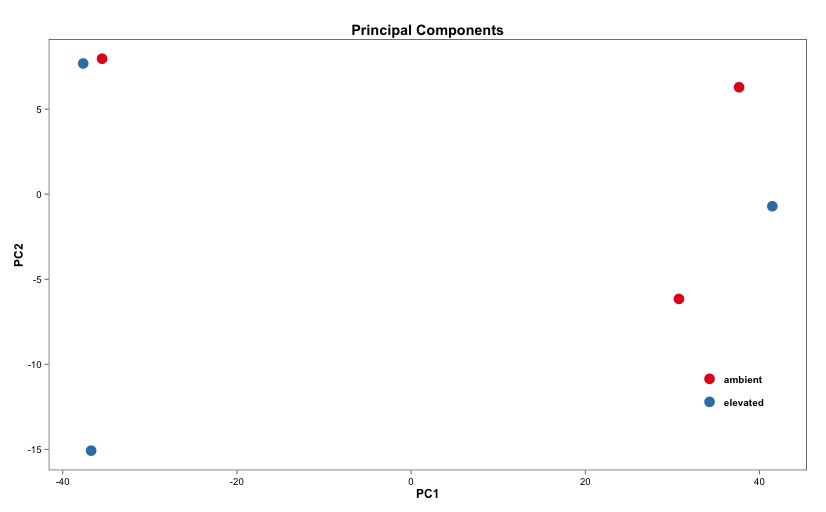
Merging Data and Using Biomart:
To continue with analysis, we can use the .csv files we generated from the DeSEQ2 analysis and find gene ontology. This next script contains the actual biomaRt calls, and uses the .csv files to search through the Phytozome database. If you are trying to search through other datsets, simply replace the “useMart()” command with the dataset of your choice. Again, the biomaRt call is relatively simple, and this script is customizable in which values you want to use and retrieve.
# Install biomaRt package source("http://bioconductor.org/biocLite.R") biocLite("biomaRt") # Load biomaRt library library("biomaRt") # Convert final results .csv file into .txt file results_csv <- "Gmax_DESeq2-replaceoutliers-results.csv" write.table(read.csv(results_csv), gsub(".csv",".txt",results_csv)) results_txt <- "Gmax_DESeq2-replaceoutliers-results.txt" #biomaRt is for http only so to get around this you need to add this line options(RCurlOptions=list(followlocation=TRUE, postredir=2L)) # List available datasets in database # Then select dataset to use ensembl = useMart("phytozome_mart", host="phytozome.jgi.doe.gov",path="/biomart/martservice", dataset="phytozome") listDatasets(ensembl) # Check the database for entries that match the IDs of the differentially expressed genes from the results file a <- read.table(results_txt, head=TRUE) b <- getBM(attributes=c('transcript_id', 'chr_name1', 'gene_name1', 'gene_chrom_start', 'gene_chrom_end', 'kog_desc'), filters=c('pac_transcript_id'), values=a$X, mart=ensembl) biomart_results <- "Gmax_DESeq2-Biomart.txt" write.table(b,file=biomart_results)
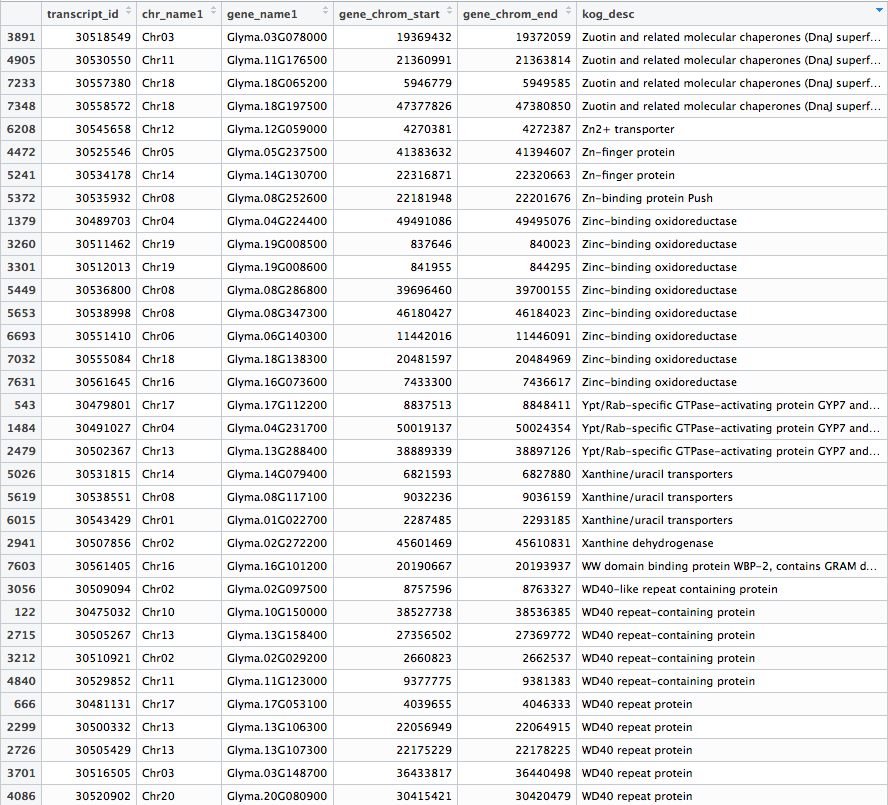
After fetching data from the Phytozome database based on the PAC transcript IDs of the genes in our samples, a .txt file is generated that should look something like this:
Finally, we want to merge the deseq2 and biomart output.
# Merge the deseq2 and biomart output m <- merge(a, b, by.x="X", by.y="transcript_id") write.csv(as.data.frame(m),file = "Gmax_DEseq2-Biomart-results_merged.csv")
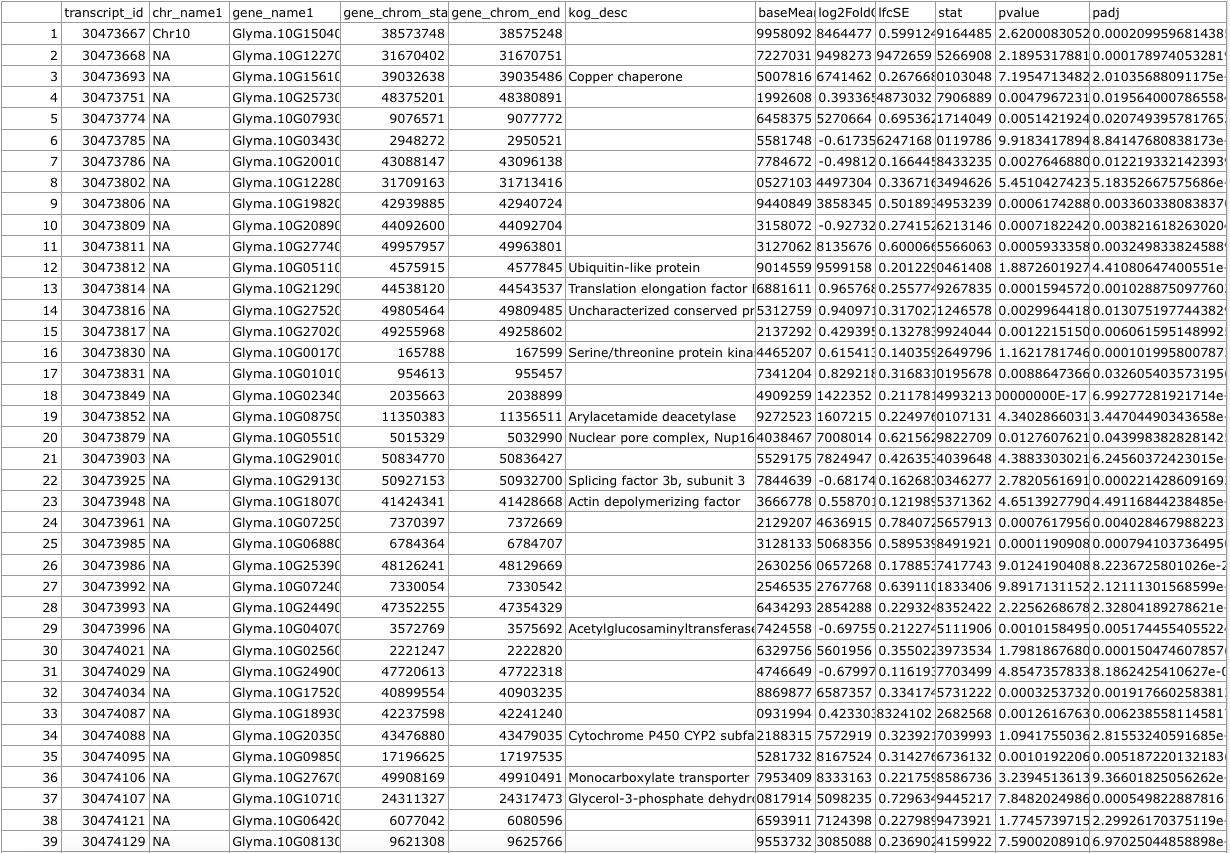
We get a merged .csv file with our original output from DESeq2 and the Biomart data:
Visualizing Differential Expression with IGV:
To visualize how genes are differently expressed between treatments, we can use the Broad Institute’s Interactive Genomics Viewer (IGV), which can be downloaded from here: IGV
We will be using the .bam files we created previously, as well as the reference genome file in order to view the genes in IGV. IGV requires that .bam files be indexed before being loaded into IGV. There is a script file located in
/common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping/bam_files called bam_index.sh that will accomplish this. The .bam files themselves as well as all of their corresponding index files (.bai) are located here as well. The reference genome file is located at
/common/RNASeq_Workshop/Soybean/gmax_genome/Gmax_275_v2
You will need to download the .bam files, the .bai files, and the reference genome to your computer. Once you have IGV up and running, you can load the reference genome file by going to Genomes -> Load Genome From File… in the top menu. Now you can load each of your six .bam files onto IGV by going to File -> Load from File… in the top menu. Be sure that your .bam files are saved in the same folder as their corresponding index (.bai) files. Once you have everything loaded onto IGV, you should be able to zoom in and out and scroll around on the reference genome to see differentially expressed regions between our six samples.
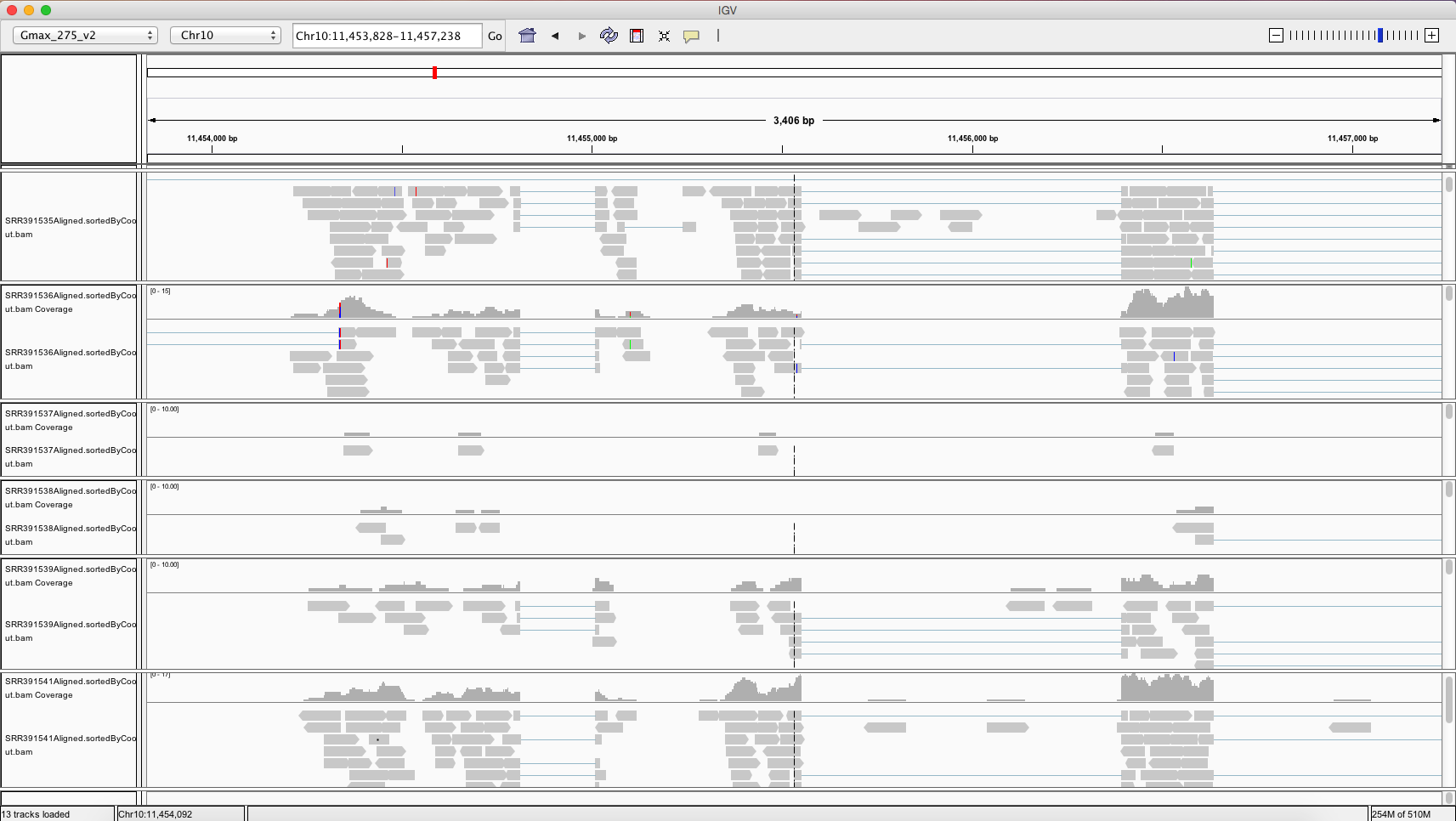
The differentially expressed gene shown is located on chromosome 10, starts at position 11,454,208, and codes for a transferrin receptor and related proteins containing the protease-associated (PA) domain. This information can be found on line 142 of our merged csv file. You can search this file for information on other differentially expressed genes that can be visualized in IGV!
Counting exon for DEXSeq:
Similar to DESeq2, DEXSeq runs statistical analysis on exon count tables generated by mapping. However, since the quantity of exon is substantial larger than that of genes, DEXSeq on a whole genome is not suitable for the tutorial in terms of runtime. In order to reduce run-time of DEXSeq , we have to divide the previously mapped BAM files by chromosome, and select one chromosome each sample for remaining tutorial:
bamtools split -in /common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping/bam_files/SRR391535Aligned.sortedByCoord.out.bam -reference
Repeat it for SRR391536, SRR391537, SRR391538, SRR391539, and SRR391541. In this tutorial, we choose chromosome 2 as our example. Scripts and output are located in
/common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping/
The Python script dexseq_prepare_annotation.py takes an Ensembl GTF file and translates it into a GFF file with collapsed exon counting bins which is required in next step.
python /opt/R/R-3.1.1/lib64/R/library/DEXSeq/python_scripts/dexseq_prepare_annotation.py /common/RNASeq_Workshop/Soybean/DEXseq/Glycine_max.V1.0.28.gtf Glycine_max.V1.0.28.gff
You can find the GTF file Glycine_max.V1.0.28.gtf from /common/RNASeq_Workshop/Soybean/DEXseq/Gmax_ensembl_genome
Next, we need to divide the reference GFF file by chromosome to match what we have done on BAM files. Because the first column of GFF file are chromosome number, we can use grep to extract chromosome 2 by searching given pattern. The result is saved into a new file called chr02.gff.
grep -w ^2 Glycine_max.V1.0.28.gff > chr02.gff
Note that the chromosome number in GFF file is marked by a single digit integer, while, in BAM files, the chromosome are marked by a string “Chr02”. In order to successfully generate reads count, we need reformat the GFF file by adding a string “Chr0” before every chromosome number at first column:
awk '{$1 = "Chr0"$1; print }' chr02.gff > chr02_mod.gff
The above command messes with the formatting of file i.e. it replaces tabs with space. The command below solves the issue and output a GFF file, called chr02_mod2.gff, which would be used in counting.
awk -v OFS="\t" '$1=$1' chr02_mod.gff > chr02_mod2.gff
In this tutorial, we choose chromosome 2 from divided BAM files for DEXSeq analysis. For each BAM file, we count the number of reads that overlap with each of the exon counting bins defined in the flattened GFF file. This is done with the script python_count.py from HTSeq package. “-f” indicates the input file is bam format. SRR391535.txt is the name of output file specified by users.
python /opt/R/R-3.1.1/lib64/R/library/DEXSeq/python_scripts/dexseq_count.py -f bam /common/RNASeq_Workshop/Soybean/DEXseq/chr02_mod2.gff /common/RNASeq_Workshop/Soybean/STAR_HTSEQ_mapping/bam_files/SRR391535/SRR391535Aligned.sortedByCoord.out.REF_Chr02.bam SRR391535.txt
This command expects two files in the current working directory, namely the GFF file produced in the previous step and a BAM file with the aligned reads from the sample. Use the script multiple times to produce a count file from each of your BAM file. Repeat this command for SRR391536, SRR391537,SRR391538, SRR391539, and SRR39154. The script and output are located in /common/RNASeq_Workshop/Soybean/DEXseq/
Analysis of Differential Exon Usage with DEXSeq:
We need to download the output file the python scirpts for DEXSeq. The R script can be found in /common/RNASeq_Workshop/Soybean/DEXseq/ Make sure the path of these files matches the path of your working directory. To install DEXSeq library in R. start R console and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("DEXSeq")
DEXSeq
In our R script, the first section should include working directory setup and file import:
directory <- "~/workspace/R/DEXSeq/" setwd(directory) countFiles = list.files(directory, pattern=".txt$", full.names=TRUE) basename(countFiles) flattenedFile = "chr02.gff" basename(flattenedFile)
Note that here we use unprocessed chr02.gff instead of chr02_mod2.gff used in mapping. Next, we need to prepare a sample table. This table should contain one row for each library, and columns for all relevant information such as name of the file with the read counts, experimental conditions, technical information and further covariates.
sampleNames <- c("Leaf tissue 1","Leaf tissue 2","Leaf tissue 3","Leaf tissue 4","Leaf tissue 5","Leaf tissue 6")
sampleCondition <- c("ambient","ambient","elevated","elevated","elevated","ambient")
samplelibType <- c("single-end", "single-end", "single-end", "single-end", "single-end", "single-end")
sampleTable <- data.frame(sampleName = sampleNames, condition = sampleCondition, libTpye = samplelibType)
Now, we construct an DEXSeqDataSet object from this data. This object holds all the input data and will be passed along the stages of the subsequent analysis. We construct the object with the DEXSeq function DEXSeqDataSetFromHTSeq, as follows:
suppressPackageStartupMessages( library( "DEXSeq" ) )
dxd = DEXSeqDataSetFromHTSeq(countFiles,
sampleData=sampleTable,
design= ~ sample + exon + condition:exon,
flattenedfile=flattenedFile)
The function takes four arguments. First, a vector with names of count files, i.e., of files that have been generated with the dexseq_count.py script. The function will read these files and arrange the count data in a matrix, which is stored in the DEXSeqDataSet object dxd. The second argument is our sample table, with one row for each of the files listed in the first argument. This information is simply stored as is in the object’s meta-data slot (see below). The third argument is a formula of the form “ sample + exon + condition:exon” that specifies the contrast with of a variable from the sample table columns and the ‘exon’ variable. Using this formula, we are interested in differences in exon usage due to the ‘condition’ variable changes. Later in this vignette, we will how to add additional variables for complex designs. The fourth argument is a file name, now of the flattened GFF file that was generated with dexseq_prepare_annotation.py and used as input to dexseq_count.py when creating the count file.
After the dxd object successfully generated, we may loading and inspect our data by using following commands.
colData(dxd) # This annotation, as well as all the information regarding each column of the DEXSeqDataSet head( counts(dxd), 5 ) # Access the first 5 rows from the count data head( featureCounts(dxd), 5 ) # Access only the first five rows from the count belonging to the exonic regions
In the following sections, we will update the object by calling a number of analysis functions, always using the idiom “dxd = someFunction( dxd )”, which takes the dxd object, fills in the results of the performed computation and writes the returned and updated object back into the variable dxd Different samples might be sequenced with different depths. In order to adjust for such coverage biases, we estimate size factors, which measure relative sequencing depth:
dxd = estimateSizeFactors( dxd )
To test for differential exon usage, we need to estimate the variability of the data. This is necessary to be able to distinguish technical and biological variation (noise) from real effects on exon usage due to the different conditions.
dxd = estimateDispersions( dxd )
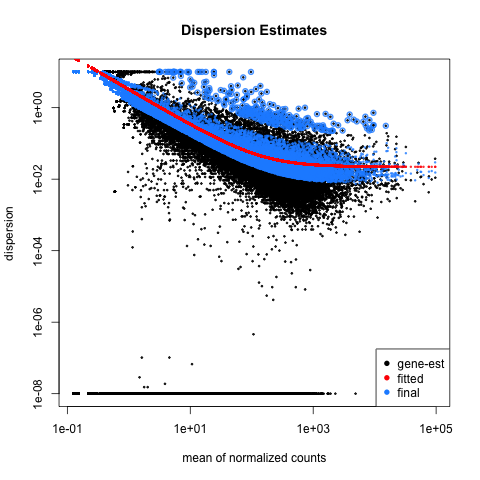
Function plotDispEsts allows us to plot the per-exon dispersion estimates versus the mean normalised count as shown below, where the initial per-exon dispersion estimates are shown by black points. shrinked values are shown in blue, the fitted mean-dispersion values are shown in red:
plotDispEsts( dxd )

Function testForDEU performs several test based on x2-distribution
dxd = testForDEU( dxd )
From the coefficients of the fitted model, it is possible to distinguish overall gene expression effects, that alter the counts from all the exons, from exon usage effects, that are captured by the interaction term condition:exon and that affect the each exon individually.
dxd = estimateExonFoldChanges( dxd, fitExpToVar="condition")
So far in the pipeline, the intermediate and final results have been stored in the meta data of a DEXSeqDataSet object, they can be accessed using the function mcol. In order to summarize the results without showing the values from intermediate steps, we call the function DEXSe qResults. The result is a DEXSeqResults object, which is a subclass of a DataFrame object.
dxr1 = DEXSeqResults( dxd ) dxr1 mcols(dxr1)$description
From this object, we can ask how many exonic regions are significant with a false discovery rate of 10%:
table ( dxr1$padj < 0.1 )
We may also ask how many genes are affected
table ( tapply( dxr1$padj < 0.1, dxr1$groupID, any ) )
To see how the power to detect differential exon usage depends on the number of reads that map to an exon, a so-called MA plot is useful. type:
plotMA( dxr1, cex=0.8 )
Below is an example output of plotMA, where red dots represents insignificant exon, here, the exons with an adjusted p values of less than 0.1,There is of course nothing special about the number 0.1, and you can specify other thresholds in the call to plotMA:
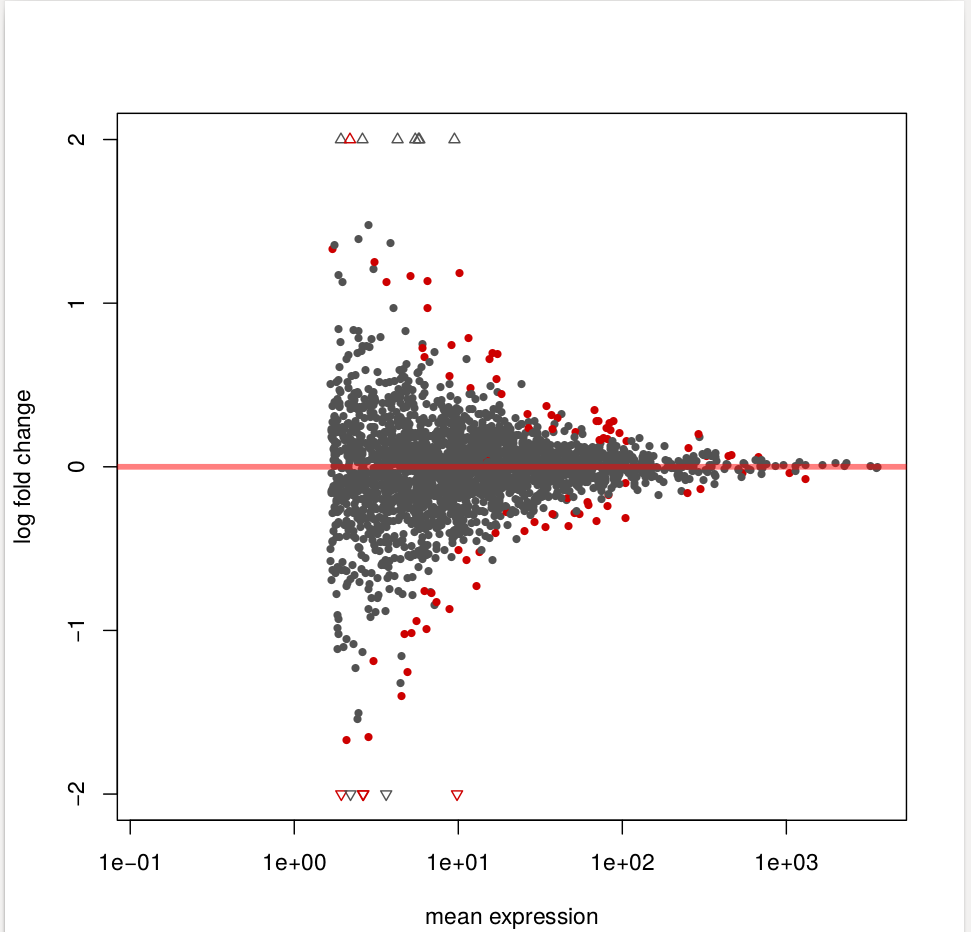
Function plotDEXSeq provides various means for us to visualize the result. The function plotDEXSeq expects at least two arguments, the DEXSeqDataSet object and the gene ID. The option legend=TRUE causes a legend to be included. The three remaining arguments in the code chunk above are ordinary plotting parameters which are simply handed over to R’s standard plotting functions. This plot shows the fitted expression values of each of the exons of gene GLYMA0002S50, where red line is exon expression under ambient condition, blue line is exon expression under elevated condition, the grey blocks are normal exons, the purple blocks represent those exons showing alternative splicing:
dxr2 = DEXSeqResults( dxd ) plotDEXSeq( dxr2, "GLYMA0002S50", legend=TRUE, cex.axis=1.2, cex=1.3, lwd=2 )

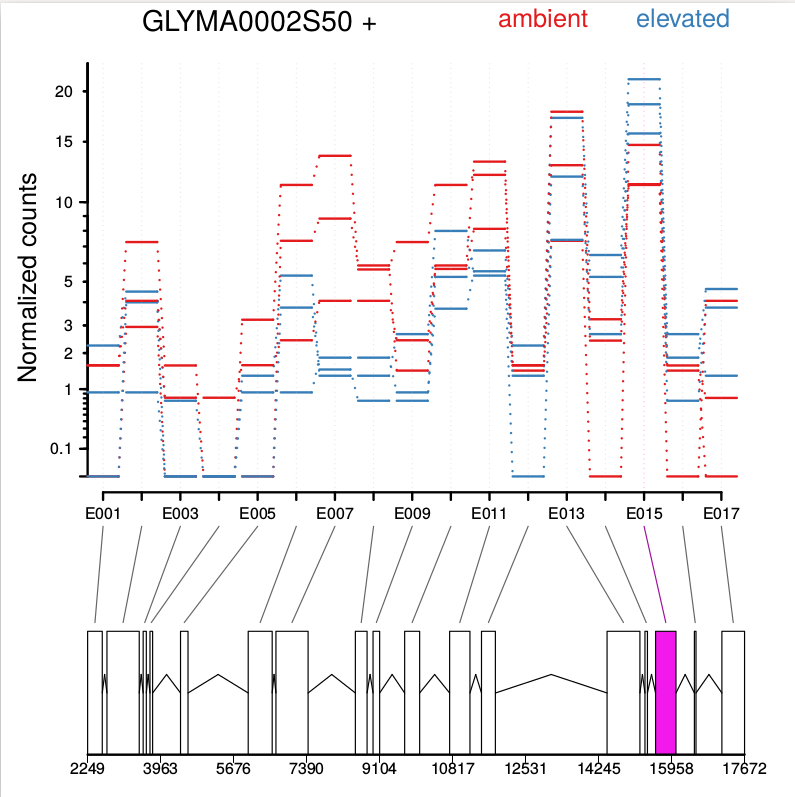
Optionally, one can also visualize the transcript models by adding displayTranscripts=TRUE, which can be useful for putting differential exon usage results into the context of isoform regulation. The graph below shows an annotated transcript model.
plotDEXSeq( dxr2, "GLYMA0002S50", displayTranscripts=TRUE, legend=TRUE, cex.axis=1.2, cex=1.3, lwd=2 )

DEXSeq is designed to find changes in relative exon usage, i. e., changes in the expression of individual exons that are not simply the consequence of overall up- or down-regulation of the gene. To visualize such changes, it is sometimes advantageous to remove overall changes in expression from the plots. The graph shows expression level of all six samples:
plotDEXSeq( dxr2, "GLYMA0002S50", expression=FALSE, norCounts=TRUE, legend=TRUE, cex.axis=1.2, cex=1.3, lwd=2 )
Function DEXSeqHTML generates an easily browsable, detailed overview over all analysis results
DEXSeqHTML( dxr2, FDR=0.1, color=c("#FF000080", "#0000FF80") )