This tutorial will serve as a guideline for RNA sequence analysis of a non-model organism. We will be using a needle transcriptome from red spruce as our example. The RNA-Seq data consists of Illumina short reads as well as 454 reads.
We will be going through:
- Quality control of the libraries
- De novo assembly of the reads
- Clustering of the assembled transcripts from each assembly
- Mapping the reads back to assembled/clustered transcriptome
- Generating raw counts
- GFOLD to analyze differential gene expression between two treatments (elevated and ambient CO2).
The packages we’ll be using can be found here:
Sickle
SolexaQA
Trinity
MIRA
Vsearch
TransDecoder
Bowtie2
eXpress
GFold
The data consists of three libraries under three different treatments;
- ambient CO2
- elevated CO2
- one that had been cotreated for both conditions
De Novo non model species directory
/common/RNASeq_Workshop/RedSpruceEach step in this work flow is composed into a directory
RedSpruce
|-- Reads
|-- QualityControl
|-- Assembly
|-- Clustered
|-- CodingRegions
|-- Index
|-- Align
|-- Count
|-- gfold
`-- blast2go
Raw Reads
Two types of sequencing methods were used to collect the RNA-Seq data, namely Illumina and 454. Where Illumina reads are in the form of fastq, where as 454 reads are in the form of fasta and quality score (qual) files.
Reads/
|-- 454/
| |-- control_454.fasta
| |-- control_454.qual
| |-- cotreated_454.fasta
| |-- cotreated_454.qual
| |-- elevated_454.fasta
| `-- elevated_454.qual
`-- Illumina/
|-- ambient.fastq
|-- cotreated.fastq
`-- elevated.fastq
Quality control
Quality control of Illumina reads using Sickle:
Step one is to perform quality control on the reads, and we will be using Sickle for the Illumina reads. Using the Sickle program we will trim our unpaired raw reads, to keep a minimum quality score value of 30 and a minimum length of 35.
sickle se -f ../../Reads/Illumina/ambient.fastq -t illumina -o ambient.trimmed.fastq -q 30 -l 35 sickle se -f ../../Reads/Illumina/elevated.fastq -t illumina -o elevated.trimmed.fastq -q 30 -l 35 sickle se -f ../../Reads/Illumina/cotreated.fastq -t illumina -o cotreated.trimmed.fastq -q 30 -l 35
The trimmed output files are what we will be using for the next steps of our analysis.
se unpaired reads / single-end reads
-f input file
-o output file
-q minimum quality score
-l minimum read length
-t Type of quality score value, e.g. (solexa (CASAVA < 1.3), illumina (CASAVA 1.3 to 1.7), sanger (which is CASAVA >= 1.8)) (required)The full script for trimming of raw reads using sickle can be found at: /common/RNASeq_Workshop/RedSpruce/QualityControl/Illumina/sickle.sh
Once the run is completed the directory will contain the following files that has been trimmed, which will be used in the further analysis:
QualityControl/
|--Illumina/
|-- ambient.trimmed.fastq
|-- cotreated.trimmed.fastq
|-- elevated.trimmed.fastq
`-- sickle_267830.out
Quality control of 454 Reads using SolexaQA:
Prior to running the quality control on the 454 reads we need to create a fastq file using our raw reads. We do have the sequence file in ‘fasta’ format and the quality score in the ‘qual’ format file. Using the python scripts (454_to_fastq.py), we will combine the sequence data and the quality score into a single file which will be a ‘fastq’ file. The newly created fastq files will be placed in the Reads directory, with the fasta and the qual files.
python 454_to_fastq_2.py ../../Reads/454/elevated_454.fasta ../../Reads/454/elevated_454.qual
python 454_to_fastq_v2.py ../../Reads/454/control_454.fasta ../../Reads/454/control_454.qual
python 454_to_fastq_v2.py ../../Reads/454/cotreated_454.fasta ../../Reads/454/cotreated_454.qual
The python script, will convert the 454 raw reads to Illumina reads (known as Solexa in the past). Where it will create fastq files (elevated_454.fastq, control_454.fastq, cotreated_454.fastq) and the new files will be written in to the /RedSpruce/Reads/454 directory folder. We have prepared a script that can be submitted using the qsub command, and it can be found at /common/RNASeq_Workshop/RedSpruce/QualityControl/454/make454_fastq.sh
Once it executes, it will create the following files in the /Reads/454 directory with the rest of the raw files:
Reads/
|-- 454/
|-- elevated_454.qual
|-- elevated_454.fastq
|-- elevated_454.fasta
|-- cotreated_454.qual
|-- cotreated_454.fastq
|-- cotreated_454.fasta
|-- control_454.qual
|-- control_454.fastq
`-- control_454.fasta
Next we use SolexaQA to perform quality control of the 454 reads. Initially we will be using the dynamictrim to trim the reads using a chosen threshold for the quality (if not defined the default value for p = 0.05). Before running the command, we will create three directories, which the trimmed reads will be copied into using the following;
#Will create the following directories in the current folder
DIR_LIST="elevated control cotreated"
for dir in $DIR_LIST; do
if [ ! -d "$dir" ]; then
mkdir -p $dir
fi
done
SolexaQA dynamictrim ../../Reads/454/elevated_454.fastq --torrent -d elevated
SolexaQA dynamictrim ../../Reads/454/control_454.fastq --torrent -d control
SolexaQA dynamictrim ../../Reads/454/cotreated_454.fastq --torrent -d cotreated
Usage: SolexaQA dynamictrim [Options]
Options Explained:
-p|--probcutoff probability value (between 0 and 1) at which base-calling error is considered too high (default; p = 0.05)
-d|--directory path to directory where output files are saved
-t|--torrent Ion Torrent fastq file
Three output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.trimmed — a FASTQ file containing the trimmed reads.
*.segments — This tab delimited text file displays the proportion of all reads that contain a segment of a given length.
*.segments.hist.pdf — a histogram showing the distribution of the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05).
Next lengthsort will be use to sort the reads based on the length. Here we use length cutoff value as 50 bp.
SolexaQA lengthsort elevated/elevated_454.fastq.trimmed -l 50
SolexaQA lengthsort elevated/elevated_454.fastq.trimmed -l 50
SolexaQA lengthsort cotreated/cotreated_454.fastq.trimmed -l 50
Usage: SolexaQA lengthsort [options]
Options explained:
-l|--length length cutoff [defaults to 25 nucleotides]
The lengthsort program will generate *.single *.discarded *.summary.txt *.summary.txt.pdf files, where the *.single file will contain the reads which passed the length cut off value we have set. If you have paired-end reads it will be sorted in to two paired read files. (more information can be found in the SolexaQA website)
Once both SolexaQA commands have been executed, it will produce the following files in the corresponding directories:
QualityControl/
|--454/
|-- control/
| |-- control_454.fastq.trimmed
| |-- control_454.fastq.trimmed.discard
| |-- control_454.fastq_trimmed.segments
| |-- control_454.fastq_trimmed.segments_hist.pdf
| |-- control_454.fastq.trimmed.single
| |-- control_454.fastq.trimmed.summary.txt
| `-- control_454.fastq.trimmed.summary.txt.pdf
|-- cotreated/
| |-- cotreated_454.fastq.trimmed
| |-- cotreated_454.fastq.trimmed.discard
| |-- cotreated_454.fastq_trimmed.segments
| |-- cotreated_454.fastq_trimmed.segments_hist.pdf
| |-- cotreated_454.fastq.trimmed.single
| |-- cotreated_454.fastq.trimmed.summary.txt
| `-- cotreated_454.fastq.trimmed.summary.txt.pdf
`-- elevated/
|-- elevated_454.fastq.trimmed
|-- elevated_454.fastq.trimmed.discard
|-- elevated_454.fastq_trimmed.segments
|-- elevated_454.fastq_trimmed.segments_hist.pdf
|-- elevated_454.fastq.trimmed.single
|-- elevated_454.fastq.trimmed.summary.txt
`-- elevated_454.fastq.trimmed.summary.txt.pdf
The resulting *.single files will be used for the next step.
We are using MIRA for the next step with the 454 trimmed reads, and the program expect the input files to have a proper extension where it can identify the data type, for that we are going to rename the *.single file to *.single.fastq files. We have included the copying step into the script, SolexaQA.sh and it is located at /common/RNASeq_Workshop/RedSpruce/QualityControl/454. Where;
for dir in $DIR_LIST; do
for f in $dir/*.single; do
cp $f "$dir/$(basename "$f").fastq"
done
doneOnce the extension has been corrected, we will be using the *.single.fastq files for the next step.
De novo assembly
In De novo assembly section, we will be woking in the /Assembly directory. Where Illumina quality controlled reads will be assembled using Trinity and the 454 quality controlled reads will be assembled using the MIRA. For each assembly there will be separate directories called /Trinity and /MIRA.
De novo assembly of Illumina reads using Trinity:
Now we assemble all three quality controlled Illumina libraries with Trinity which is a de novo transcriptome assembler. This is most appropriate to use on a read set where no reference genome exists or where the reference genome is not high quality. Assembly requires a great deal of memory (RAM) and can take a few days to run if the read set is large. The BBC cluster has a high memory node you can connect to access this. In this example, we are assembling short reads using the highmem.q which has a 514 GB RAM. The script trinity.sh can be found at /common/RNASeq_Workshop/RedSpruce/Assembly/Trinity.
module load trinity/2.4.0
Trinity --seqType fq \
--max_memory 256G \
--single ../../QualityControl/Illumina/ambient.trimmed.fastq,../../QualityControl/Illumina/cotreated.trimmed.fastq,../../QualityControl/Illumina/elevated.trimmed.fastq \
--min_contig_length 300 \
--CPU 16 \
--normalize_reads
If you do not specify a output directory, the out put files will be directed to the trinity default folder called trinity_out_dir/.
Assembly/
|--trinity_out_dir/
|-- chrysalis/
|-- inchworm.K25.L25.DS.fa
|-- inchworm.K25.L25.DS.fa.finished
|-- inchworm.kmer_count
|-- insilico_read_normalization/
|-- jellyfish.kmers.fa
|-- jellyfish.kmers.fa.histo
|-- output_tutorial.txt
|-- partitioned_reads.files.list
|-- partitioned_reads.files.list.ok
|-- pipeliner.35650.cmds
|-- read_partitions/
|-- recursive_trinity.cmds
|-- recursive_trinity.cmds.completed
|-- recursive_trinity.cmds.ok
|-- single.fa
|-- single.fa.ok
|-- single.fa.read_count
`-- Trinity.fasta
There will be number of files created and the assembled file will be called Trinity.fasta. We will be using this for our next step.
To check the quality of your Trinity assembly we can use a utility program provided with the trinity software called TrinityStats.pl. To look at the contig summery we use a script called “trinity_stats.sh” located at, /common/RNASeq_Workshop/RedSpruce/Assembly/Trinity in the cluster.
perl $TRINITY_HOME/util/TrinityStats.pl trinity_out_dir/Trinity.fasta > TrinitySummary.txtIf you submit the using a script, the result will be piped in to the output file call “TrinitySummary.txt” and it will look like:
################################
## Counts of transcripts, etc.
################################
Total trinity 'genes': 63856
Total trinity transcripts: 114946
Percent GC: 42.14
########################################
Stats based on ALL transcript contigs:
########################################
Contig N10: 3636
Contig N20: 2760
Contig N30: 2230
Contig N40: 1835
Contig N50: 1492
Median contig length: 662
Average contig: 1022.21
Total assembled bases: 117498506
#####################################################
## Stats based on ONLY LONGEST ISOFORM per 'GENE':
#####################################################
Contig N10: 3583
Contig N20: 2710
Contig N30: 2170
Contig N40: 1761
Contig N50: 1389
Median contig length: 582
Average contig: 940.82
Total assembled bases: 60076924
De novo assembly of 454 reads using MIRA:
Trinity is specialized for short read data so different assemblers will be used for the Roche 454. In this case, we will use MIRA [MIRA documentation] which also requires substantial memory and potentially time to complete the assemly. All three quality controlled 454 libraries will be assembled togehter.
In order to run mira, one must first construct a configuration file or a so called manifest file (Mira_454.conf), which is given bellow for this exercise. The configuration file contains two parts; The First part contains the general information, and the Second part contains information about the sequencing data.
The script can be found in the following path : /common/RNASeq_Workshop/RedSpruce/Assembly/Mira
# First part: defining some basic things
# We just give a name to the assembly : Mira_454
# and tell MIRA it should assemble a EST/RNASeq de-novo in accurate mode
# As special parameters, we want to use 8 threads (not : number of threads) in parallel
project = Mira_454
job = est,denovo,accurate
parameters = -GE:not=8
#The second part defines the sequencing data MIRA should load and assemble,
#and the data is logically divided into readgroups
readgroup = Control_454
data = /Common/RNASeq_Workshop/RedSpruce/QualityControl/454/control/control_454.fastq.trimmed.single.fastq
technology = 454
readgroup = Elevated_454
data = /Common/RNASeq_Workshop/RedSpruce/QualityControl/454/elevated/elevated_454.fastq.trimmed.single.fastq
technology = 454
readgroup = Cotreated_454
data = /Common/RNASeq_Workshop/RedSpruce/QualityControl/454/cotreated/cotreated_454.fastq.trimmed.single.fastq
technology = 454
For the MIRA run, the files need to be loaded into a local disk space in the cluster. Depending on the queue you are using in BBC, you can assign two different locations for the data to be loaded into.
all.q /scratch
highmem.q /scratch OR /scratch2
For the purpose of this run we will define a temporary space, called WORKING_DIR pointing to local location in the cluster. So we will load the data to this space, and once the run is completed we are going to copy the data back to our destination directory, which will be defined by the DESTINATION_DIR.
Following script mira_454.sh will be located at: /common/RNASeq_Workshop/RedSpruce/Assembly/Mira
module load mira/4.9.5
WORKING_DIR="/scratch/$USER/Mira/est"
if [ ! -d "$WORKING_DIR" ]; then
mkdir -p $WORKING_DIR
fi
DESTINATION_DIR="/common/RNASeq_Workshop/RedSpruce/Assembly/Mira"
cd $WORKING_DIR
cp $DESTINATION_DIR/* .
mira Mira_454.conf > Mira_454_log.txt
cp -r * $DESTINATION_DIR
rm -rf $WORKING_DIR
MIRA assembly run will create a directory called Mira_454_assembly and will contain unto four sub-directories.
Mira_454_d_results: This will have the output files of the assembly in different formats
Mira_454_d_info: This will contain the information and statics files of the final assembly
Mira_454_d_tmp: Contains tmp files and temporary assembly files
Mira_454_d_chkpt: This contain the checkpoint files needed to resume the job if it crashed or stopped
The Mira_454_d_info directory will contain the assembled files in fasta format.
Assembly/
|-- Mira/
|-- Mira_454.conf
|-- mira_454.sh
|-- Mira_454_assembly/
| |-- Mira_454_d_chkpt/
| |-- Mira_454_d_info/
| |-- Mira_454_d_results/
| | |-- Mira_454_out.caf
| | |-- Mira_454_out.maf
| | |-- Mira_454_out.padded.fasta
| | |-- Mira_454_out.padded.fasta.qual
| | |-- Mira_454_out.tcs
| | |-- Mira_454_out.txt
| | |-- Mira_454_out.unpadded.fasta
| | `-- Mira_454_out.unpadded.fasta.qual
| `-- Mira_454_d_tmp/
`-- Mira_454_log.txt
MIRA will write two kind to fasta files: padded and unpadded. The padded format will contain the gap information in the form of asterisks and in the unpadded version these gaps will be removed. For our next step we will be using the unpadded version (Mira_454_out.unpadded.fasta).
Clustering
Clustering of Illumina and 454 reads using vsearch:
To obtain a set of unique genes from both runs, we will cluster the two resulting assemblies together. First, the two assembies will be combined into one file using the Unix command cat, which refers to concatanate.
cat ../Assembly/Trinity/trinity_out_dir/Trinity.fasta ../Assembly/Mira/Mira_454_assembly/Mira_454_d_results/Mira_454_out.unpadded.fasta > combine.fasta Once the files are combined, we will use vsearch to find redundancy between the assembled transcripts and create a single output known as a centroids file. The threshold for clustering in this example is set to 80% identity. The following script combine.sh will be located at /common/RNASeq_Workshop/RedSpruce/Clustered
vsearch --threads 8 --log LOGFile \
--cluster_fast combine.fasta \
--id 0.80 --centroids centroids.fasta --uc clusters.uc
Usage: vsearch [OPTIONS]
--threads INT number of threads to use, zero for all cores (0)
--log FILENAME write messages, timing and memory info to file
--cluster_fast FILENAME cluster sequences after sorting by length
--id REAL reject if identity lower, accepted values: 0-1.0
--centroids FILENAME output centroid sequences to FASTA file
--uc FILENAME specify filename for UCLUST-like output
After the run, it will contain the following files; where the centroids.fasta file will contain the unique genes from the 454 and Illumina assemblies.
Clustered/
|-- centroids.fasta
|-- clusters.uc
|-- combine.fasta
|-- combine.sh
`-- LOGFile
For the next step we will be using the centroids.fasta file.
Identifying the Coding Regions
Identifying coding regions using TransDecoder:
Now that we have our reads assembled and clustered together into the single centroids file, we can use TransDecoder to determine optimal open reading frames from the assembly (ORFs). Assembled RNA-Seq transcripts may have 5′ or 3′ UTR sequence attached and this can make it difficult to determine the CDS in non-model spieces.
First we will use the TransDecoder utility to run on the centroid fasta file, as follows:
TransDecoder.LongOrfs -t ../Clustered/centroids.fasta
Usage: Transdecoder.LongOrfs [Options]
Options explained:
-t transcripts.fastaBy default it will identify ORFs that are at least 100 amino acids long. (you can change this by using -m parameter). It will produce a folder called centroids.fasta.transdecoder_dir, and will include all the temporary files in that directory.
CodingRegions/
|-- centroids.fasta.transdecoder_dir/
|-- longest_orfs.pep
|-- longest_orfs.gff3
|-- longest_orfs.cds
|-- base_freqs.dat.ok
`-- base_freqs.dat
Next step is to, identify ORFs with homology to known proteins via blast or pfam searches. This will maximize the sensitivity for capturing the ORFs that have functional significance. It done using the following code:
hmmscan --cpu 8 --domtblout pfam.domtblout /common/Pfam/Pfam-B.hmm centroids.fasta.transdecoder_dir/longest_orfs.pep
It will create a file called pfam.domtblout in the main directory.
The final step is to predict the likely coding regions using the Pfam (or Blast) search results, using the following:
TransDecoder.Predict -t ../Clustered/centroids.fasta --retain_pfam_hits pfam.domtblout --cpu 8
The full script (Transdecoder.sh) for identifying the coding regions using the TransDecoder is located at: /common/RNASeq_Workshop/RedSpruce/CodingRegions/Transdecoder.sh
After the run is completed it will create the following files:
CodingRegions/
|-- centroids.fasta.transdecoder_dir/
| |-- longest_orfs.pep
| |-- longest_orfs.gff3.inx
| |-- longest_orfs.gff3
| |-- longest_orfs.cds.top_longest_5000.nr80.clstr
| |-- longest_orfs.cds.top_longest_5000.nr80
| |-- longest_orfs.cds.top_longest_5000
| |-- longest_orfs.cds.top_500_longest.ok
| |-- longest_orfs.cds.top_500_longest
| |-- longest_orfs.cds.scores.selected
| |-- longest_orfs.cds.scores.ok
| |-- longest_orfs.cds.scores
| |-- longest_orfs.cds.eclipsed_removed.gff3
| |-- longest_orfs.cds.best_candidates.gff3
| |-- longest_orfs.cds
| |-- hexamer.scores.ok
| |-- hexamer.scores
| |-- base_freqs.dat.ok
| `-- base_freqs.dat
|-- pfam.domtblout
|-- centroids.fasta.transdecoder.pep
|-- centroids.fasta.transdecoder.gff3
|-- centroids.fasta.transdecoder.cds
`-- centroids.fasta.transdecoder.bed
In the next step, for creating an index, we will be using the centroids.fasta.transdecoder.cds file.
Creating A Index
Indexing the reads using bowtie2:
Before aligning the reads we need to create a index for the created transcoder assembly. We will be using Bowtie2 to build the index using the following command:
bowtie2-build ../CodingRegions/centroids.fasta.transdecoder.cds centroids_transdecoder_Index
Usage: bowtie2-build [options] [reference_in] [bt2_index_base]
main arguments
reference_in comma-separated list of files with ref sequences
bt2_index_base base name for the index files to write
Options
--threads # of threads, By default bowtie2-build is using only one thread. Increasing the number of threads will speed up the index building considerably in most cases.The script (bowtie2.sh) for creating the index can be found at /common/RNASeq_Workshop/RedSpruce/Index/bowtie2.sh
Index/
|-- centroids_transdecoder_Index.1.bt2
|-- centroids_transdecoder_Index.2.bt2
|-- centroids_transdecoder_Index.3.bt2
|-- centroids_transdecoder_Index.4.bt2
|-- centroids_transdecoder_Index.rev.1.bt2
`-- centroids_transdecoder_Index.rev.2.bt2
Aligning the Reads to the Index
Aligning the reads using bowtie2:
Once the index has been created, we will use bowtie2 to align our three trimmed reads, using the following command:
bowtie2 --threads 8 -x ../Index/centroids_transdecoder_Index ../QualityControl/Illumina/elevated.trimmed.fastq -S elevated.sam
bowtie2 --threads 8 -x ../Index/centroids_transdecoder_Index ../QualityControl/Illumina/ambient.trimmed.fastq -S ambient.sam
bowtie2 --threads 8 -x ../Index/centroids_transdecoder_Index ../QualityControl/Illumina/cotreated.trimmed.fastq -S cotreated.sam
Usage: bowtie2 [Options] -x [bt2_base_index] {reads / options}
Main arguments:
-x [bt2_base_index] The basename of the index for the reference genome (i.e. with out the *.bt2 extension)
-S Output file name of the SAM alignment
Options:
--threads Number of processors
The script bowtie2_Align.sh used to align using the bowtie2 can be found at /common/RNASeq_Workshop/RedSpruce/Align/. Once aligned it will produce three SAM files.
Align/
|-- ambient.sam
|-- cotreated.sam
`-- elevated.sam
Converting SAM to BAM format:
SAM format file is a large human readable file. In a computational point of view, accessing this data for processing will take time. For faster processing this need to be converted into computer friendly binary format called the BAM files. We use samtools to convert the SAM file in to BAM files. Using the view command the it will be converted into BAM format and will be sorted using the sort command as shown bellow:
samtools view -uhS elevated.sam | samtools sort - elevated_sorted
samtools view -uhS ambient.sam | samtools sort - ambient_sorted
samtools view -uhS cotreated.sam | samtools sort - cotreated_sorted
Usage: samtools [command] [options] in.sam
Command:
view prints all alignments in the specified input alignment file (in SAM, BAM, or CRAM format) to standard output in SAM format
Options:
-h Include the header in the output
-S Indicate the input was in SAM format
-u Output uncompressed BAM. This option saves time spent on compression/decompression and is thus preferred when the output is piped to another samtools command
Usage: samtools [command] [-o out.bam]
Command:
sort Sort alignments by leftmost coordinates
-o Write the final sorted output to FILE, rather than to standard output.
The script (sam2bam.sh) can be found at /common/RNASeq_Workshop/RedSpruce/Align. Once the conversion is done it will produce the following files which will be binary format.
Align/
|-- elevated_sorted.bam
|-- cotreated_sorted.bam
`-- ambient_sorted.bam
Aligned Read Counts
Extracting Raw Read Counts with eXpress:
Once it has been aligned, we will use the eXpress software, to get the counts of differential expression.
express ../CodingRegions2/centroids.fasta.transdecoder.cds ../Align/elevated_sorted.bam -o elevated_express
express ../CodingRegions2/centroids.fasta.transdecoder.cds ../Align/ambient_sorted.bam -o ambient_express
express ../CodingRegions2/centroids.fasta.transdecoder.cds ../Align/cotreated_sorted.bam -o cotreated_express
Usage: express [options] [target_seqs.fa] [hits.(sam/bam)]
Required arguments:
target_seqs.fa target sequence file in fasta format
hits.(sam/bam) read alignment file in SAM or BAM format
Options:
-o [ --output-dir ] arg (=.) write all output files to this directory
The script (express.sh) can be found at /common/RNASeq_Workshop/RedSpruce/Count/express.sh directory. The program will run and will create a directory for each run and will include the output files inside the directories.
Count\
|-- elevated_express\
| |-- results.xprs
| `-- params.xprs
|-- cotreated_express\
| |-- results.xprs
| `-- params.xprs
`-- ambient_express\
|-- results.xprs
`-- params.xprs
Analysis
Converting counts to Gfold format:
Before analyzing for the differential expression using the gfold package, we need to convert our counts to Gfold count format. The program expects, 5 data columns, which are “Gene Symbol”, “Gene Name”, “Read Count”, “Gene exon length”, “FPKM”. To do this we will read the results.xprs file and grab the columns which have the required fields, and will rewrite them to a new file, where it will be used as the input file for the Gfold program.
require("plyr")
express_file_1 <- data.frame(read.table("../Count/ambient_express/results.xprs",sep="\t", header = TRUE))
express_file_2 <- data.frame(read.table("../Count/cotreated_express/results.xprs",sep="\t", header = TRUE))
express_file_3 <- data.frame(read.table("../Count/elevated_express/results.xprs",sep="\t", header = TRUE))
subset1 <- data.frame(express_file_1$target_id, express_file_1$target_id, express_file_1$tot_counts, express_file_1$length,express_file_1$fpkm)
subset2 <- data.frame(express_file_2$target_id, express_file_2$target_id, express_file_2$tot_counts, express_file_2$length,express_file_2$fpkm)
subset3 <- data.frame(express_file_3$target_id, express_file_3$target_id, express_file_3$tot_counts, express_file_3$length,express_file_3$fpkm)
sorted1 <- subset1[order(express_file_1$target_id),]
sorted2 <- subset2[order(express_file_2$target_id),]
sorted3 <- subset3[order(express_file_3$target_id),]
subset1 <- rename(subset1,c("express_file_1.target_id"="GeneSymbol","express_file_1.target_id.1"="GeneName","express_file_1.tot_counts"="Read Count","express_file_1.length" ="Gene_exon_length","express_file_1.fpkm"="RPKM"))
subset2 <- rename(subset2,c("express_file_2.target_id"="GeneSymbol","express_file_2.target_id.1"="GeneName","express_file_2.tot_counts"="Read Count","express_file_2.length" ="Gene_exon_length","express_file_2.fpkm"="RPKM"))
subset3 <- rename(subset3,c("express_file_3.target_id"="GeneSymbol","express_file_3.target_id.1"="GeneName","express_file_3.tot_counts"="Read Count","express_file_3.length" ="Gene_exon_length","express_file_3.fpkm"="RPKM"))
write.table(sorted1, "ambient.read_cnt", sep = "\t", row.names = FALSE, col.names = FALSE, quote = FALSE)
write.table(sorted2, "cotreated.read_cnt", sep = "\t", row.names = FALSE, col.names = FALSE, quote = FALSE)
write.table(sorted3, "elevated.read_cnt", sep = "\t", row.names = FALSE, col.names = FALSE, quote = FALSE)
We have created a R script (eXpress2gfold.R), which will be called, through eXpress2gfold.sh script. The script is located at /common/RNASeq_Workshop/RedSpruce/gfold/eXpress2gfold.sh .
The above script will produce the following gfold read count files as the output, which will be used in analyzing the differential expression.
gfold/
|-- ambient.read_cnt
|-- cotreated.read_cnt
`-- elevated.read_cnt
Differential expression using Gfold:
Using the formatted count files, we will use Gfold to analyze the differential expression of the three samples. Gfold program is very useful when no replicates are available. Gfold generalizes the fold change by, considering the log fold change value for each gene, and assigns it as a Gfold value. It overcomes the short comping of the p-value, that measure the expression under different conditions, by measuring a relative expression value. It also overcomes the deficiency of low read counts. The following code finds the differentially expressed genes between the two groups of samples.
gfold diff -s1 cotreated -s2 elevated -suf .read_cnt -o cotreated_VS_elevated.diff
gfold diff -s1 cotreated -s2 ambient -suf .read_cnt -o cotreated_VS_ambient.diff
gfold diff -s1 elevated -s2 ambient -suf .read_cnt -o elevated_VS_ambient.diffUsage: gfold [jobs] [options]
Jobs:
diff Calculate GFOLD value and other statics. It accepts the count values as input.
Options explained:
-s1 prefix for read_count of 1st group separated by comma.
-s2 prefix for read_count of 2nd group separated by comma.
-suf suffix of count file
-o output file and for diff job, there will be two output files, where second will have .ext extension.
-norm normalization method. 'Count' stands for normalization by total number of mapped reads.
The prepared script for running Gfold is called, gfold.sh and it is located at: /common/RNASeq_Workshop/RedSpruce/gfold. The job will create following files:
gfold/
|-- cotreated_VS_ambient.diff
|-- cotreated_VS_ambient.diff.ext
|-- cotreated_VS_elevated.diff
|-- cotreated_VS_elevated.diff.ext
|-- elevated_VS_ambient.diff
`-- elevated_VS_ambient.diff.ext
The first few lines of the output file of cotreated_VS_ambient.diff will look like:
# This file is generated by gfold V1.1.4 on Wed Jul 19 13:51:02 2017
# Normalization constants :
# cotreated 52838582 1.0984
# elevated 55466744 1
# The GFOLD value could be considered as a reliable log2 fold change.
# It is positive/negative if the gene is up/down regulated.
# A gene with zero GFOLD value should never be considered as
# differentially expressed. For a comprehensive description of
# GFOLD, please refer to the manual.
#GeneSymbol GeneName GFOLD(0.01) E-FDR log2fdc 1stRPKM 2ndRPKM
Gene.10065 Gene.10065 -0.15334 1 -0.2063 84.977 63.876
Gene.10096 Gene.10096 2.46607 1 2.5501 25.495 129.567
Gene.10097 Gene.10097 1.90102 1 2.072 27.710 101.209
Gene.10138 Gene.10138 1.38044 1 1.4319 55.704 130.366
Gene.10198 Gene.10198 -0.24752 1 -0.2928 121.876 86.279
.
.
.
Gene.10423 Gene.10423 2.83385 1 2.91202 26.908 175.725
Gene.10440 Gene.10440 0 1 -0.05898 55.542 46.239
Gene.10487 Gene.10487 2.39166 1 2.56462 7.236 37.198
The output file contains 7 columns, namely “GeneSymbol”, “GeneName”, “GFOLD(0.01)”,
“E-FDR”, “log2fdc”, “1stRPKM”, “2ndRPKM”. From that, we will focus on the GFOLD column for each gene. The GFOLD value, can be considered as a reliable log2 fold value indicating the change. The positive value indicate a up regulated and a negative value indicate a down regulated gene.
Functional annotation of assembled transcripts with BLAST2GO:
Before proceed to the Functional annotation, we need to select the most differential expressed genes from GFOLD output and extract corresponded nucleotide sequences as input file of Blast2go. Python scirpt ExtractSeqence.py will do the job. The script takes three arguments, a “diff” file generated from GFOLD, a f”asta” file containning either protein sequences or nucleotide sequences, and a number n indicating first n gene IDs users want to select. The script ranks genes in diff file based on GFOLD value. In this tutorial, the first 5 Gene IDs are selected to extract sequences from the cds file. 5 non-identical Sequences are successfully found and stored in a output fasta file.
python ExtractSequence.py ../gfold/cotreated_VS_ambient.diff ../CodingRegions/centroids.fasta.transdecoder.cds 5The Script ExtractSequence.py can be found at /common/RNASeq_Workshop/RedSpruce/blast2go/. The directory after running is shown below:
blast2go/
|-- ExtractSequence.py
`-- ExtractedSq.fasta
Blast2GO is an interactive bioinformatics platform for functional annotation and analysis of genomic datasets. Blast2GO incorporates NCBI BLAST and various public databases to identify sequences. It also allows user to easily visualize sequenced genomes and associated functional interpretation. Its interactive interface makes it a great tool for starter in bioinformatics. To download and intall free version Blast2GO, go to its official website and follow the instruction. In this tutorial, we will perform function annotation on assembled transcripts that found to be deferentially expressed between treatments. We are going to run 4 analyses on the sequences: blast, mapping, annotation, interproscan (only on the PFam database) and graphs. The general work flow is shown figure below.
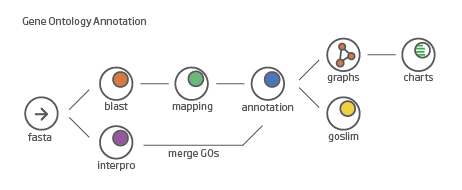
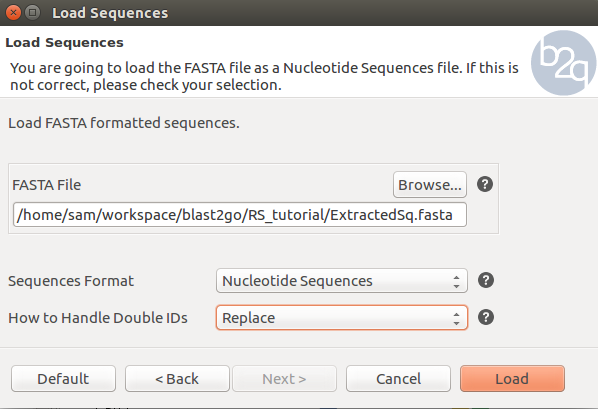
To load the output fasta file, Copy seqs_to_blast.fasta over to your local machine and open Blast2Go. Navigate to the File menu then –> Load –> Load Sequences –> Create a new project and select your fasta file. Blast2Go should create a nice table from the file.
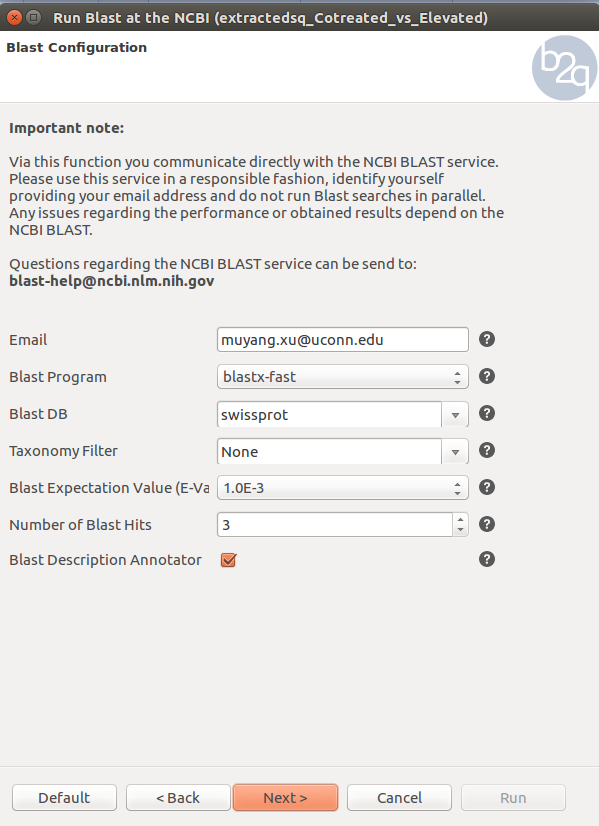
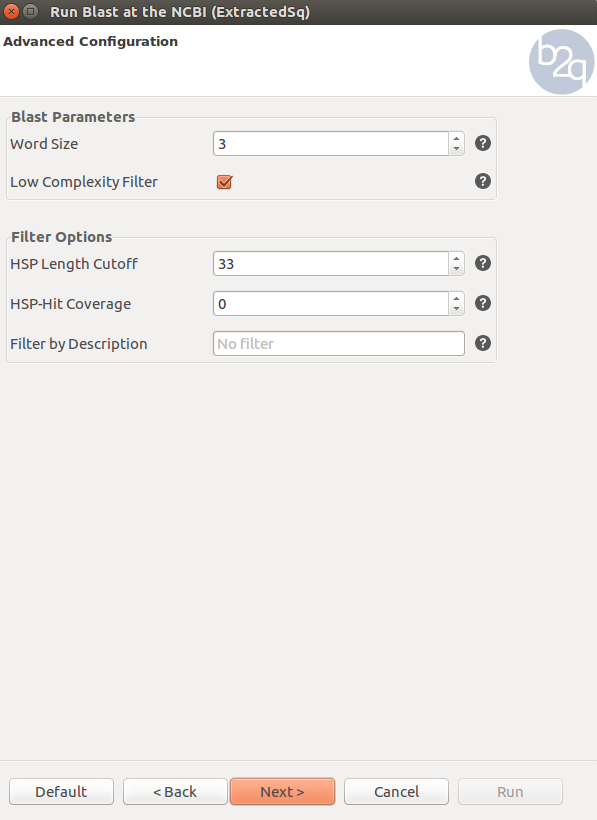
Basic Local Alignment Search Tool(BLAST) is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA sequences. A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold. In this tutorial, We are going to run NCBI blast. Make sure all sequences are selected. The blast setting are shown below. Since we use nucleotide sequence of a non-model species in this example, Blastx and swiss_prot data base are selected in order to retrieve more accurate results from query. Click question mark after each parameter to learn details of each setting. Successfully blasted sequences are labeled in pink color. Check your BLAST settings based on figures below. The blast run will take about one hour. You can access results from BLAST run is located in /common/RNASeq_Workshop/RedSpruce/blast2go/results/
Once the blast is done you can look at the BLAST results. Right click on the orange color finished sequences, and select “Show Blast Results” to show the individual result for each sequence.

For each individual BLAST result, information on the e-value, UniProt codes and more information is given.
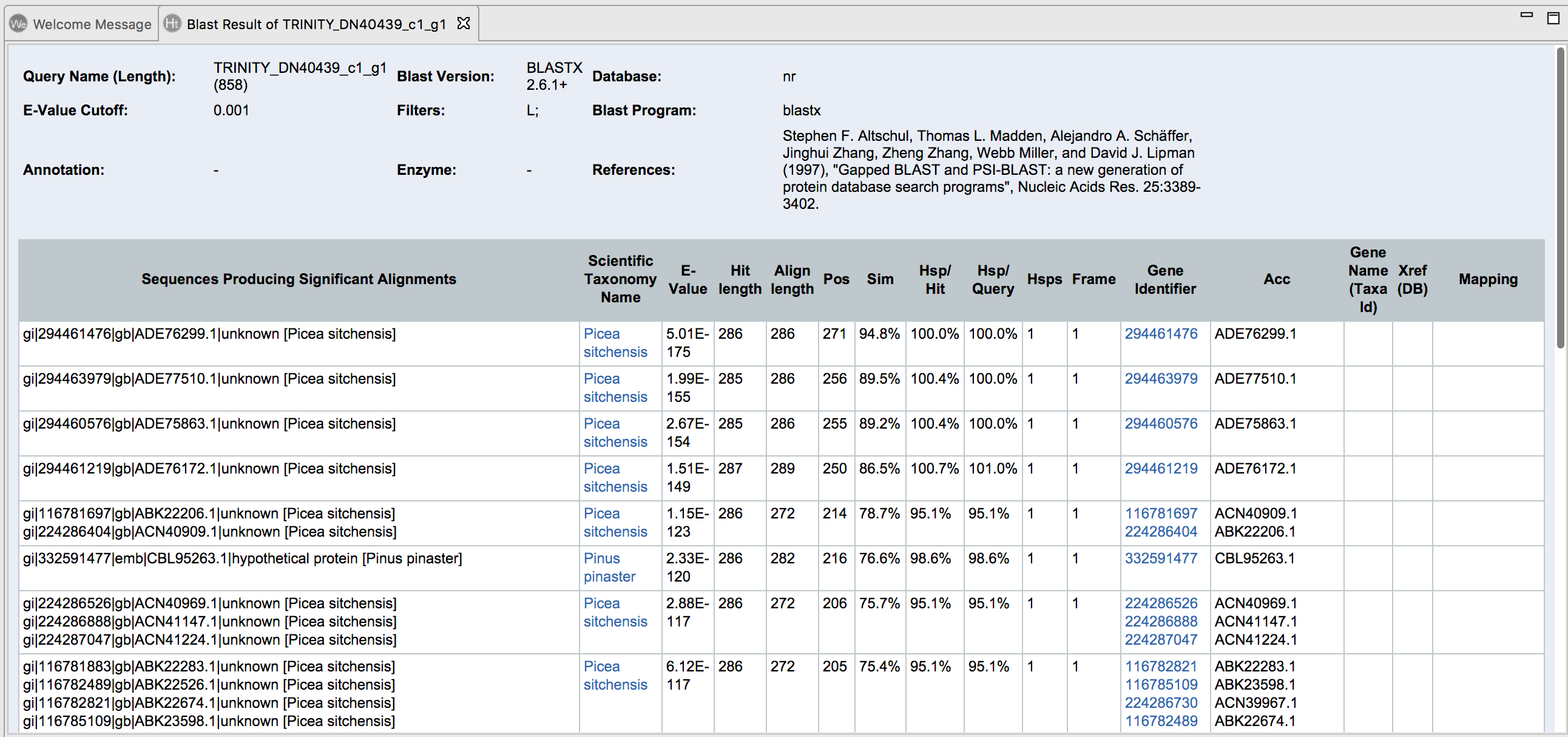
InterPro provides a one-stop-shop for protein classification, where all the signatures produced by the different member databases are placed into entries within the InterPro database. Signatures which represent equivalent domains, sites or families are put into the same entry and entries can also be related to one another. Additional information such as a description, consistent names and Gene Ontology (GO) terms are associated with each entry, where possible. To perform InterPro, click the purple symbol on the top and select only Pfam on the pop-up screen. This step can be executed concurrently with Blast, the previous step.
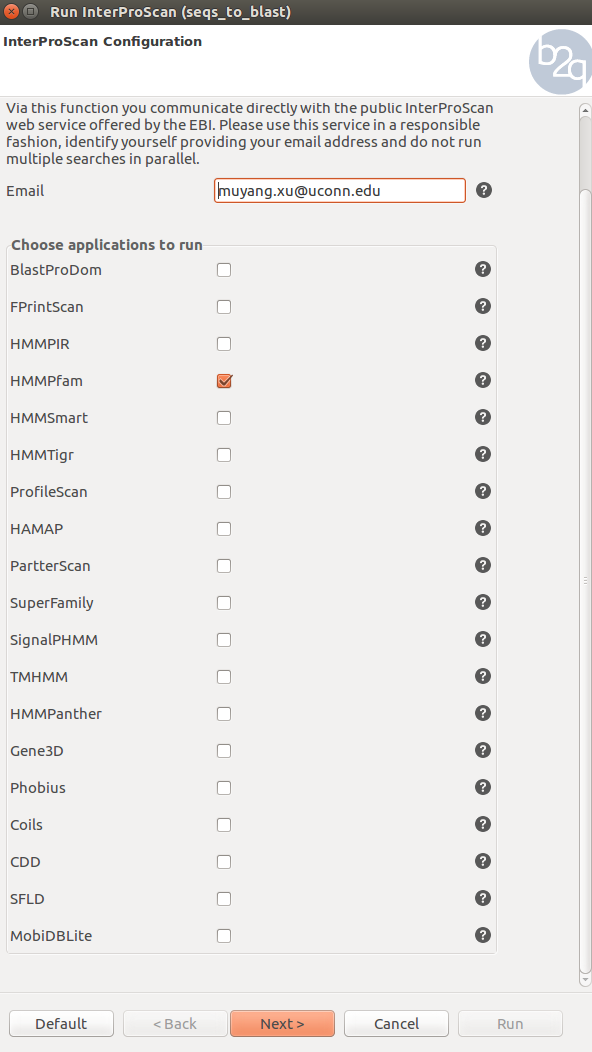
Blast2go performs different mapping steps to link all Blast Hits to the functional information stored in the Gene Ontology database. The go database contains several million functional annotated gene products for hundreds of different species. All annotations are associated to an evidence code which provides information about the quality of this functional assignment. Click the green symbol and hit run to perform mapping. Next step is annotation, where , keep the default settings and run it. Successfully mapped and annotated sequences will turn blue.
![]()
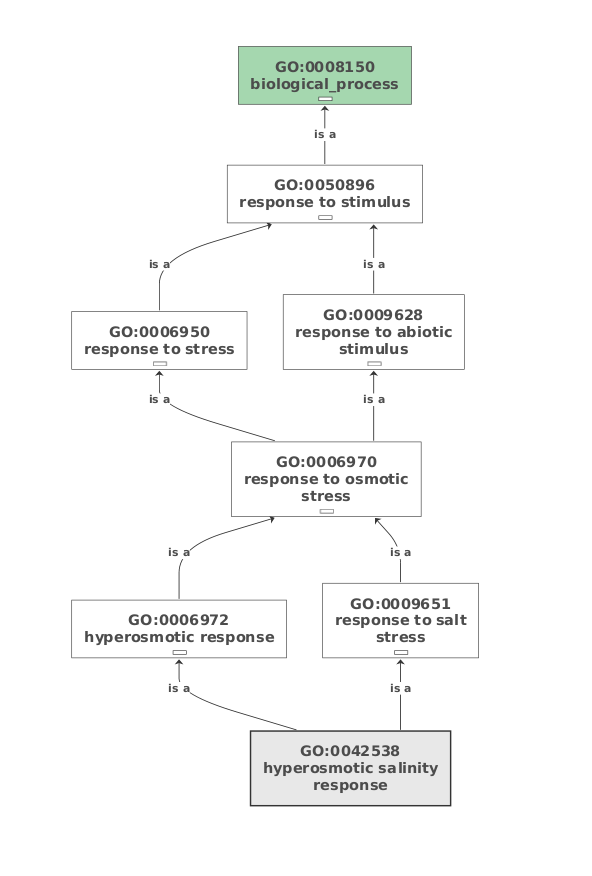
To check detailed results of each step, right click the sequence that you selected. For mapped and annotated sequences, we can visualize biological process for each GO term, right click selected sequence -> show GO description -> Choose a GO term -> click show. Figure below shows biological process generated from GO:008150 of sequence Gene.26346.
The results will help us to understand functionality of this deferentially expressed gene set. The b2g file after running is located at /common/RNASeq_Workshop/RedSpruce/blast2go/results/