This tutorial will introduce Single-cell RNA library preparation and provide guideline for single cell library analysis by using Cell Ranger. We will learn basics of Single Cell 3’ Protocol, and run Cell Ranger pipelines on a single library as demonstration.
We will go through:
The sample data in this tutorial are listed below:
- cellranger-tiny-bcl-1.2.0
- cellranger-tiny-bcl-samplesheet-1.2.0.csv
- hgmm_100_fastqs.tar
- refdata-cellranger-hg19-and-mm10-1.2.0
Library Preparation
Step 1 – GEM Generation & Barcoding
The Single Cell 3’ Protocol upgrades short read sequencers to deliver a scalable microfluidic platform for 3’ digital gene expression profiling of 500 – 10,000 individual cells per sample. The 10xTM GemCodeTM Technology samples a pool of ~ 750,000 barcodes to separately index each cell’s transcriptome. It does so by partitioning thousands of cells into nanoliter-scale Gel Bead-In-EMulsions (GEMs), where all generated cDNA share a common 10x Barcode. Libraries are generated and sequenced from the cDNA and the 10x Barcodes are used to associate individual reads back to the individual partitions.
To achieve single cell resolution, the cells are delivered at a limiting dilution, such that the majority (~90- 99%) of generated GEMs contains no cell, while the remainder largely contain a single cell.
Upon dissolution of the Single Cell 3’ Gel Bead in a GEM, primers containing (i) an Illumina R1 sequence (read 1 sequencing primer), (ii) a 16 bp 10x Barcode, (iii) a 10 bp randomer and (iv) a poly-dT primer sequence are released and mixed with cell lysate and Master Mix. Incubation of the GEMs then produces barcoded, full- length cDNA from poly-adenylated mRNA. After incubation, the GEMs are broken and the pooled fractions are recovered.
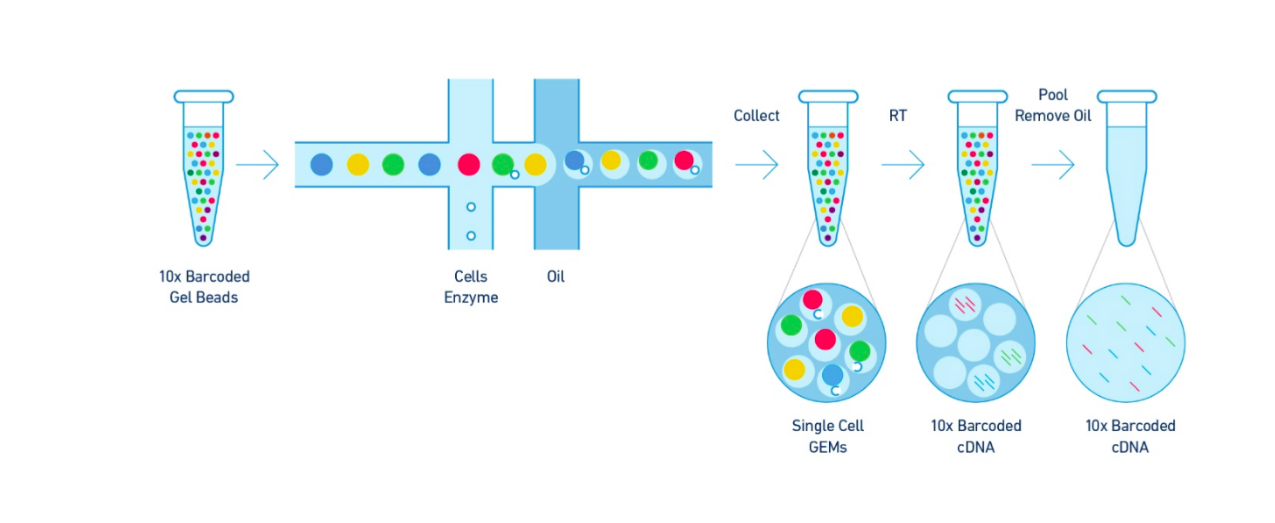
Step 2 – Post GEM-RT Cleanup & cDNA Amplification
Silane magnetic beads are used to remove leftover biochemical reagents and primers from the post GEM reaction mixture. Full-length, barcoded cDNA is then amplified by PCR to generate sufficient mass for library construction.
Step 3 – Library Construction
Enzymatic Fragmentation and Size Selection are used to optimize the cDNA amplicon size prior to library construction. R1 (read 1 primer sequence) are added to the molecules during GEM incubation. P5, P7, a sample index and R2 (read 2 primer sequence) are added during library construction via End Repair, A- tailing, Adaptor Ligation and PCR. The final libraries contain the P5 and P7 primers used in Illumina bridge amplification.
Step 4 – Sequencing Libraries
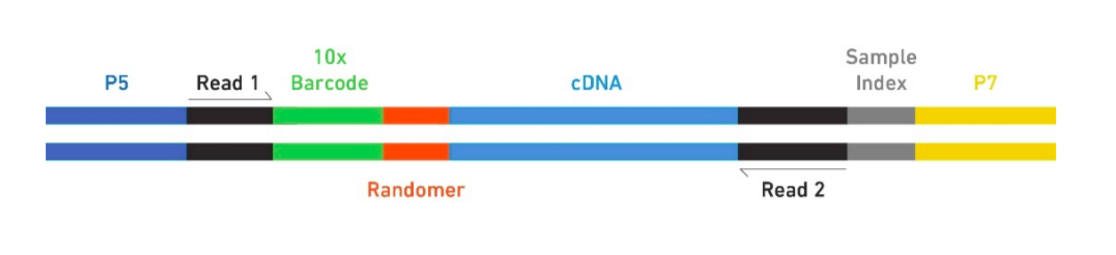
The Single Cell 3’ Protocol produces Illumina-ready sequencing libraries. A Single Cell 3’ Library comprises standard Illumina paired-end constructs which begin and end with P5 and P7. The Single Cell 3’ 16 bp 10xTM Barcode and 10 bp randomer is encoded in Read 1, while Read 2 is used to sequence the cDNA fragment. Sample index sequences are incorporated as the i7 index read. Read 1 and Read 2 are standard Illumina sequencing primer sites used in paired-end sequencing. The final structure of library are shown below, in which 10X barcode specifies cell and randomer specifies transcript.
Library Analysis
Workflows
The Cell Ranger workflow always starts with running cellranger mkfastq on each flowcell. The subsequent steps vary depending on how many samples, libraries and flowcells you have. We will describe them in order of increasing complexity:
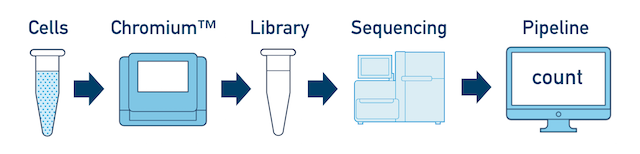
Single Sample, Library, and Flowcell is the most basic case. You have a single biological sample, which was prepared into a single library, and then sequenced on a single flowcell. Assuming the FASTQs have been generated with cellranger mkfastq, you just need to run cellranger count as described in Single-Library Analysis.
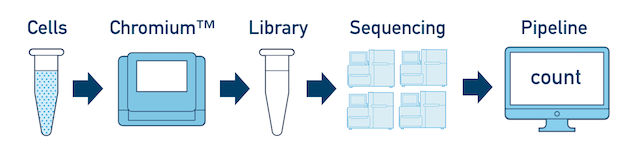
One Library, Multiple Flowcells If you have a library which was sequenced across multiple flowcells, you can pool the reads from both sequencing runs. Follow the steps in Multi-Flowcell Samples to combine them in a single cellranger count run.
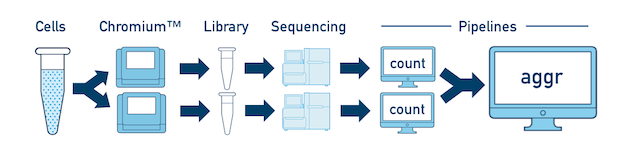
One Sample, Multiple Libraries If you prepared multiple libraries from the same sample (technical replicates, for example), then each one should be run through a separate instance of cellranger count. Once those are completed, you can perform a combined analysis using cellranger aggr, as described in Multi-Library Aggregation.
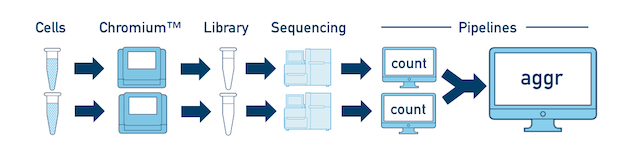
Multiple Biological Samples For a full experiment involving multiple biological samples, you must run cellranger count separately for each individual library deriving from each of those samples. For instance, if your experiment involves four samples, each having two libraries / replicates, then you will have to run cellranger count eight times. Then you can combine them all in a single call to cellranger aggr.
For the rest of this tutorial, we will go through Single Sample, Library, and Flowcell by applying mkfastq and count on two data set respectively.
The cellranger mkfastq pipeline is a 10x-enhanced wrapper around Illumina bcl2fastq, which demultiplexes BCL files from a sequencer into FASTQs for analysis. In this section, we uses the tiny-bcl example sequencing run as example. you don’t have to download file to the working directory since the file is also located at /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/, but, for teaching purpose, download instruction for sample data are shown below:
wget http://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-1.2.0.tar.gz
The file is a zipped tar file. To unzip the file, type:
tar -xvzf cellranger-tiny-bcl-1.2.0.tar.gz
This will create a new subdirectory called cellranger-tiny-bcl-1.2.0. To run mkfastq pipeline, an Illumina Experiment Manager (IEM) sample sheet is also required. Note that his sample sheet is an example that is only valid for the Single Cell 3′ v2 chemistry. enter command below to download the data sheet. The sample sheet can also be found at /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/.
wget http://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-samplesheet-1.2.0.csv
Let’s briefly look at the tiny-bcl sample sheet before running the pipeline. It is a sample sheet that in the Illumina Experiment Manager (IEM) format. Note that you can specify a 10x sample index set in the index column of the Data section:
| [Data] | |||
| Lane | Sample_ID | Index | Sample_Project |
| 5 | Sample1 | SI-GA-CS | tiny_bcl |
Here, SI-GA-C5 refers to a set of four separate sample indices. cellranger mkfastq also supports oligo sequences in the index column. In this example, only reads from lane 5 will be used. To search for the given sample index across all lanes, omit the lanes column entirely.
Before running the pipeline, we need to load cellrange by applying module load. You can check available version of Cellranger by typing module avail on Xanadu. In this tutorial, we use CellRanger/1.2.1.
module load CellRanger/1.2.1
Next, we run Cellranger to generate FASTQs. Option --run specifies path to the unzipped BCL file you want to demultiplex. Option --samplesheet specifies path sample sheet, cellranger-tiny-bcl-samplesheet-1.2.0.csv in this case.This well take a few minutes. The shell script mkfastq.sh that runs the example can be found at /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/.
cellranger mkfastq --run=/path/to/tiny_bcl \
--samplesheet=tiny-bcl-samplesheet.csvOnce the cellranger mkfastq pipeline has successfully completed, the output can be found in a new directory named with the serial number of the flowcell processed by cellranger mkfastq. The flowcell serial number for the tiny-bcl dataset is H77WWBBXX. The bcl2fastq output can be found in outs/fastq_path, and is organized in the same manner as a conventional bcl2fastq run.
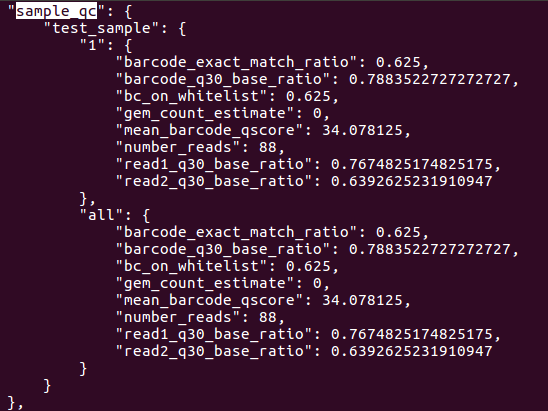
In addition to generating FASTQs, the cellranger mkfastq pipeline writes both sequencing and 10x-specific quality control metrics into a JSON file. The metrics are in the outs/qc_summary.json file.There are quite a few metrics, but a few are particularly useful. Let’s look at the sample_qc key. To read entry associated with specific key, first open the JSON file with less:
less /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/H35KCBCXY/outs/qc_summary.json
Then forward search the key by typing text below in less.
/sample_qc
This should return result as shown below. The sample_qc metric is a dictionary that contains one entry per distinct sample in the sample sheet, and one metrics structure per lane per sample, plus an ‘all’ structure in case a sample spans multiple lanes.
There are some other metrics we can check in order to diagnose low barcode mapping rates and read quality before running a cellranger pipeline. Keys and their functions are shown in chart below:
For a complete list of command-line arguments and additional information, run cellranger mkfastq --help.
cellranger count takes FASTQ files from cellranger mkfastq and performs alignment, filtering, and UMI counting. It uses the Chromium cellular barcodes to generate gene-barcode matrices and perform clustering and gene expression analysis. count can take input from multiple sequencing runs on the same library.
We will use a data set generated from 100 1:1 Mixture of Fresh Frozen Human (HEK293T) and Mouse (NIH3T3) Cells. The data set is available at /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/hgmm100. In addition, we also need a transcriptome reference datasets and run cellranger. Shell script count.sh for this pipeline can be found at /UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell.
cellranger count --cells=100 --id=hgmm --transcriptome=/UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/refdata-cellranger-hg19-and-mm10-1.2.0 --fastqs=/UCHC/PublicShare/CBC_Tutorials/Tutorials/SingleCell/hgmm100/fastqs/ --sample=read
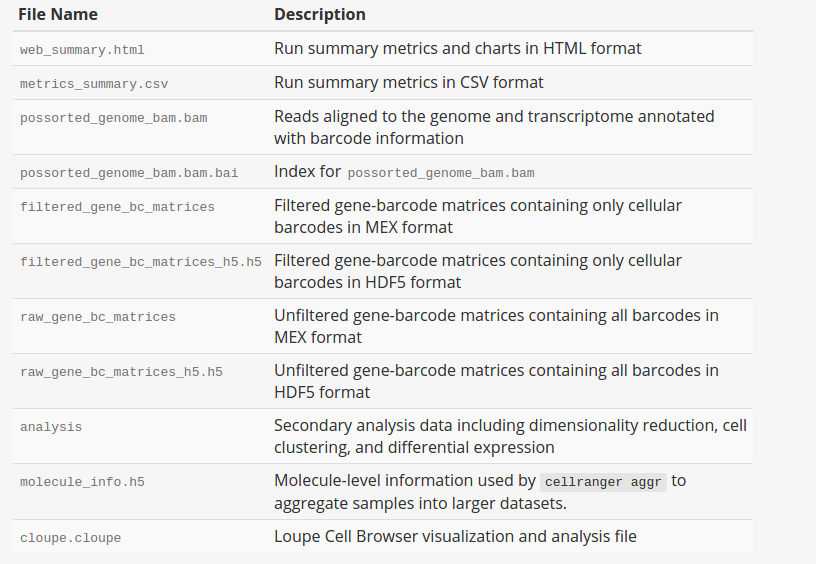
As shown above, cellranger count takes 4 required arguments. --id is a unique run ID string, which in the example is assigned arbitrarily to “hgmm”. --fastqs specifies path of the FASTQ directory that usually generated from mkfastq. Here we use fastq files of human-mouse cell mix. --sample indicates sample name as specified in the sample sheet supplied to mkfastq, which, in this case, is “hgmm100”. --transcriptom specifies path to the Cell Ranger compatible transcriptome reference, Here we use refdata-cellranger-hg19-and-mm10-1.2.0, a reference for a human and mouse mixture sample. --cells is optional flag where we can specify number of cells within the sample. The default value is 3000. For more optional arguments and additional information, enter cellranger count --help. The output of the pipeline will be contained in a diectory named with the sample ID you specified (e.g. hgmm). The subdirectory named “outs” will contain the main pipeline output files. Detailed description of output is shown chart below:
Once cellranger count has successfully completed, you can browse the resulting summary HTML file in any supported web browser, user can also open the .cloupe file in Loupe Cell Browser, or refer to the Understanding Output section to explore the data by hand. Figure below shows run summary. The summary metrics describe sequencing quality and various characteristics of the detected cells. The number of cells detected, the mean reads per cell, and the median genes detected per cell are prominently displayed near the top of the page. The Barcode Rank Plot under the “Cells” dashboard shows the distribution of barcode counts and which barcodes were inferred to be associated with cells. The y-axis is the number of UMI counts mapped to each barcode and the x-axis is the number of barcodes below that value. A steep drop-off is indicative of good separation between the cell-associated barcodes and the barcodes associated with empty partitions.
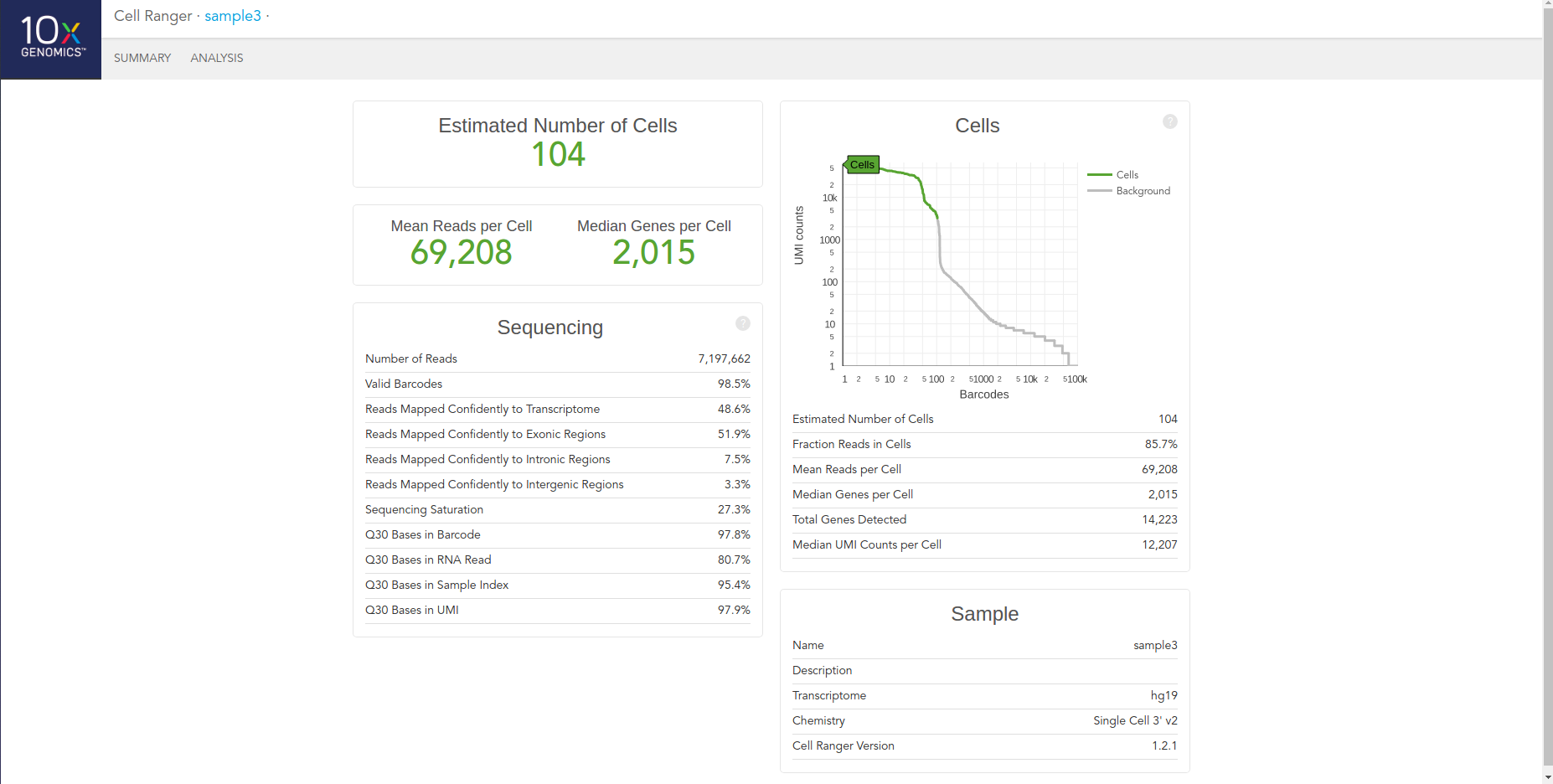
The automated secondary analysis results can be viewed by clicking “Analysis” in the top left corner. The secondary analysis provides the following:
- A dimensional reduction analysis which projects the cells into a 2-D space (t-SNE)
- An automated clustering analysis which groups together cells that have similar expression profiles
- A list of genes that are differentially expressed between the selected clusters
- A plot showing the effect of decreased sequencing depth on observed library complexity
- A plot showing the effect of decreased sequencing depth on Median Genes Detected
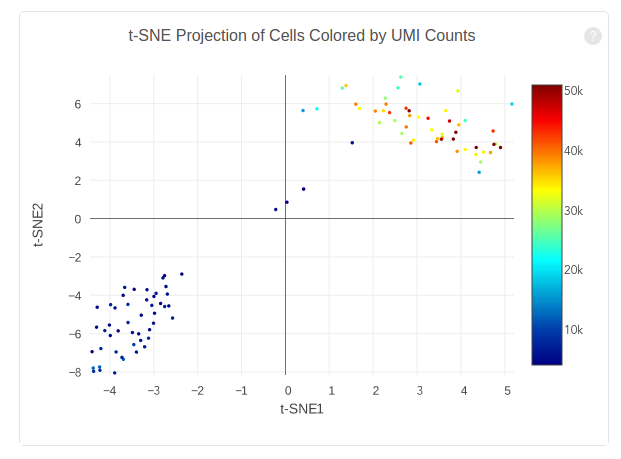
The plot below shows the 2-D t-SNE projection of the cells colored by the total UMI counts per cell. This is suggestive of the RNA content of the cells and often correlates with cell size – redder points are cells with more RNA in them. In this space, pairs of cells that are close to each other have more similar gene expression profiles than cells that are distant from each other. In his case, cells on upper left corner belong to mouse. Cells on lower right corner belong to human.
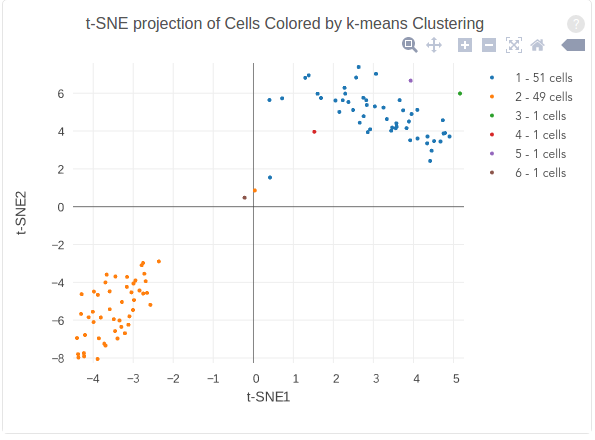
The figure below shows 2-D t-SNE projection of cells. The type of clustering analysis is selectable from the dropdown in the upper right – change this to vary the type of clustering and/or number of clusters that are assigned to the data.
The bottom left plot shows the effect of decreased sequencing depth on Sequencing Saturation, which is a measure of the fraction of library complexity that was observed. The far right point of the line is the full sequencing depth obtained in this run. Similarly, the bottom right plot shows the effect of decreased sequencing depth on Median Genes per Cell, which is a way of measuring data yield as a function of depth. The far right point is the full sequencing depth obtained in this run. Similarly, the bottom right plot shows the effect of decreased sequencing depth on Median Genes per Cell, which is a way of measuring data yield as a function of depth. The far right point is the full sequencing depth obtained in this run.

The primary analysis output of Cell Ranger is a gene-barcode matrix. Cell Ranger R Kit is an R package for secondary analysis of this matrix data, including PCA and t-SNE projection, and k-means clustering. In this section, we will run Cell Ranger R Kit on output from previous section.
To download and install the package on your local machine, type script below in R console:
source("http://cf.10xgenomics.com/supp/cell-exp/rkit-install-1.1.0.R")
Once R Kit is successfully installed, load the cellrangerRkit library in R.
library(cellrangerRkit)
Before starting the analysis, we need to load these pipeline results into your local R environment. You can load the pipeline data by specifying path to your cellranger count result as follows.
cellranger_pipestance_path <- "/path/to/your/pipeline/output/directory" gbm <- load_cellranger_matrix(cellranger_pipestance_path) analysis_results = load_cellranger_analysis_results(cellranger_pipestance_path)
The variable gbm is an object based on the Bioconductor ExpressionSet class that stores the barcode filtered gene expression matrix and metadata, such as gene symbols and barcode IDs corresponding to cells in the data set. The gene expression values, gene information and cell barcodes can be accessed using:
exprs(gbm) # expression matrix fData(gbm) # data frame of genes pData(gbm) # data frame of cell barcodes
We can access the t-SNE projection and plot the cells colored by UMI counts as follows:
tsne_proj <- analysis_results$tsne
visualize_umi_counts(gbm,tsne_proj[c("TSNE.1","TSNE.2")],limits=c(3,4),marker_size=0.5)

Instead of using raw UMI counts for downstream differential gene analysis, we will filter unexpressed genes, normalize the UMI counts for each barcode, and use the log-transformed gene-barcode matrix. After transformation, the gene-barcode matrix contains 14233 genes for 104 cells.
use_genes <- get_nonzero_genes(gbm) gbm_bcnorm <- normalize_barcode_sums_to_median(gbm[use_genes,]) gbm_log <- log_gene_bc_matrix(gbm_bcnorm,base=10) print(dim(gbm_log))
One way to identify different cell types is by expression of specific genes in cells. You can visualize expression values of a set of genes to identify cells where particular genes are up-regulated. Here, we show how to simultaneously plot the expression of 6 gene markers, one for each subplot using the visualize_gene_markers function. The gene markers are selected from scroll list of cellranger count run.
genes <- c("S100A6","S100A4","DLK1","HIST3H2A","EFEMP2","ACTA2")
tsne_proj <- analysis_results$tsne
visualize_gene_markers(gbm_log,genes,tsne_proj[c("TSNE.1","TSNE.2")],limits=c(0,1.5))

Because the input matrix gbm_log is already normalized and log10-transformed, the color indicates the UMI counts for each gene under log10 scale.
For some data sets, de novo cell-type discovery may be desired as prior knowledge of relevant marker genes can be limited or one may not want to introduce biases. In these cases it is useful to use unsupervised analysis to first algorithmically specify groupings of cells using the full data set. In our implementation, for instance, we apply k-means clustering on the top 10 principle components of the gene expression matrix (after log-transformation, centering and scaling). Because k-means clustering requires a specified number of clusters in the data set but the number of clusters in a data set may not be known a priori, it is helpful to consider multiple values of k. The output data from Cell RangerTM includes the pre-computed cluster labels sweeping k from 2 to 10. So you can quickly visualize results for different values of k and pick the one that agrees with your intuition. Here each cell is colored by its cluster ID.
n_clu <- 2:10
km_res <- analysis_results$kmeans # load pre-computed kmeans results
clu_res <- sapply(n_clu, function(x) km_res[[paste(x,"clusters",sep="_")]]$Cluster)
colnames(clu_res) <- sapply(n_clu, function(x) paste("kmeans",x,sep="."))
visualize_clusters(clu_res,tsne_proj[c("TSNE.1","TSNE.2")])
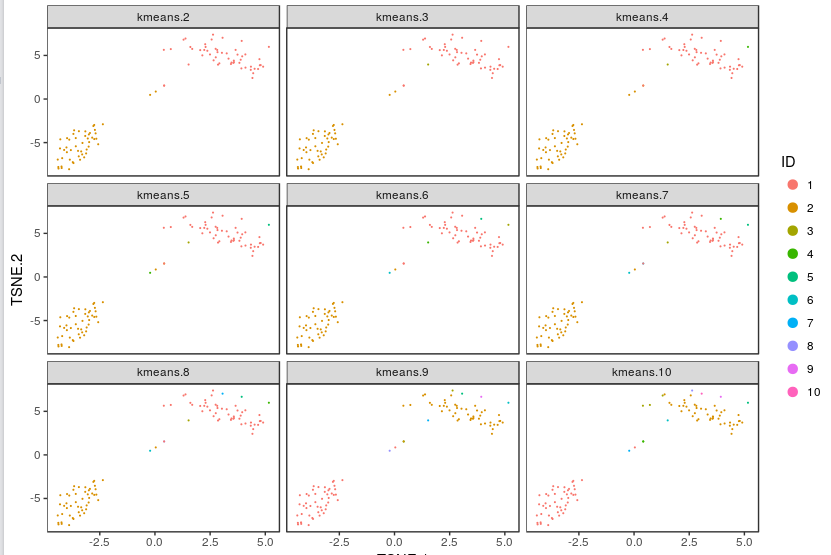
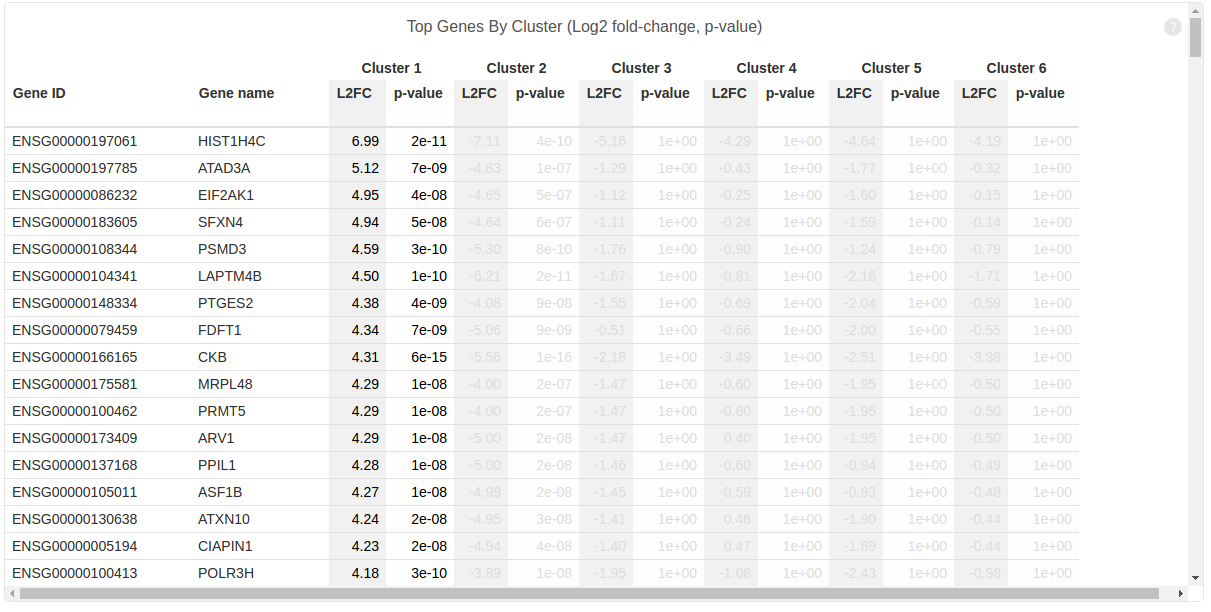
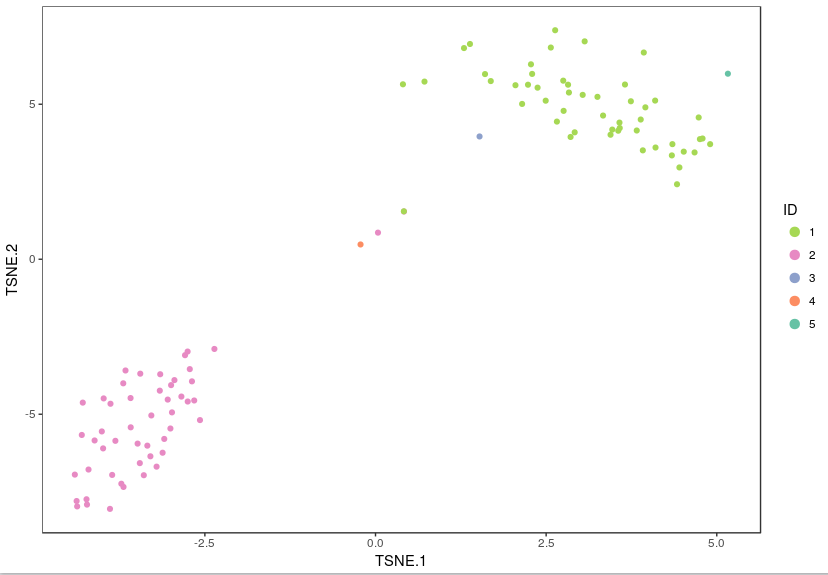
Using these integer cluster labels (or integer labels generated by any clustering algorithm), you can now perform differential gene analysis to identify gene markers that are specific to a particular cell population. For this example, we focus on the k-means clustering result above with 5 clusters. Standard statistical tests can be applied to identify which genes are most differentially expressed across different cell types.
example_K <- 5 # number of clusters (use "Set3" for brewer.pal below if example_K > 8)
example_col <- rev(brewer.pal(example_K,"Set2")) # customize plotting colors
cluster_result <- analysis_results$kmeans[[paste(example_K,"clusters",sep="_")]]
visualize_clusters(cluster_result$Cluster,tsne_proj[c("TSNE.1","TSNE.2")],colour=example_col, marker_size=1.5)
We can compare the mean expression between a class of cells and the remaining ones, and then prioritize genes by how highly expressed they are in the class of interest. The function prioritize_top_genes identifies markers that are up-regulated in particular clusters of cells.
# sort the cells by the cluster labels cells_to_plot <- order_cell_by_clusters(gbm, cluster_result$Cluster) # order the genes from most up-regulated to most down-regulated in each cluster prioritized_genes <- prioritize_top_genes(gbm, cluster_result$Cluster, "sseq", min_mean=0.5)
Now that the genes for each cell type are prioritized, you can output the top genes specific to each cluster to a local folder. In this case, we output all the top 10 gene symbols for the 5 clusters to file.
output_folder <-"/path_to_your_local_folder/pbmc_data_public/pbmc3k/gene_sets" write_cluster_specific_genes(prioritized_genes, output_folder, n_genes=10)
In addition, you can use the prioritized genes to plot a heat-map where the top three most up-regulated genes in each cluster are displayed. In addition, you can order the cells according to the cell IDs produced by k-means clustering to make the cell population signatures more apparent.
gbm_pheatmap(log_gene_bc_matrix(gbm), prioritized_genes, cells_to_plot,
n_genes=3, colour=example_col, limits=c(-1,2))
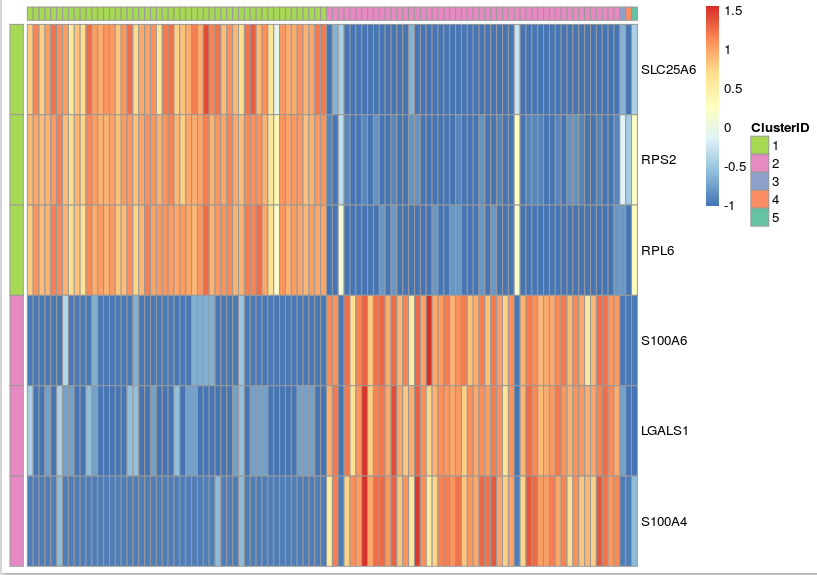
Note that, in heatmap above, Cluster 1 is labeled green and Cluster 2 is labeled purple. Gene “SLC25A6″,”RPS2″,”RPL6” are mostly expressed in Cluster 1, while “S100A6″,”LGALS1″,”S100A4” are most expressed in Cluster 2, which suggests that Cluster 1 and Cluster 2 represent mouse and human cells respectively. Therefore we can annotate the result:
cell_composition(cluster_result$Cluster,
anno=c("mouse","human","unknown1","unknowed2","unknowed3"))
Cell composition:
1 2 3 4 5
annotation "human" "mouse" "unknown1" "unknowed2" "unknowed3"
num_cells "51" "50" "1" "1" "1"
proportion "0.49038" "0.48077" "0.00962" "0.00962" "0.00962"
Reference
- Cell RangerTM R Kit Tutorial: Secondary Analysis on 10x GenomicsTM Single Cell 3’ RNA-seq PBMC Data. (n.d.). 10x Genomics. Retrieved July 11, 2017, from http://cf.10xgenomics.com/supp/cell-exp/cellrangerrkit-PBMC-vignette-knitr-1.1.0.pdf
- What is Cell Ranger (2016, November 21). Retrieved July 11, 2017, from https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger