What is Galaxy?
Galaxy is an open source, web-based platform for data intensive biomedical research. This tutorial is a transcribed version of this video tutorial from the Galaxy wiki.
Importing sample data
In this tutorial we are repeating the steps of a typical RNA-seq analysis described by Trapnell et al. (2012) with one little exception: we have created a set of smaller input files to make this tutorial faster. This data can be accessed here as a Galaxy history. Click import history and use it as a starting point of your analysis.
The data corresponds to two experimental conditions – Condition 1 and Condition 2 – each containing three replicates. In turn each replicate was sequenced as a mate-pair library and so has two associated datasets: forward and reverse. Thus (2 conditions) x (3 replicates) x (forward and reverse reads) = 2 x 3 x 2 = 12 datasets.
Creating collections of paired-end reads
First we will create a collection of the condition 1 reads. Click the Operations on multiple datasets button and check boxes will appear next to each file. Mark each of the files beginning with “c1”, then click the For all selected button and choose the Build List of Dataset Pairs function.
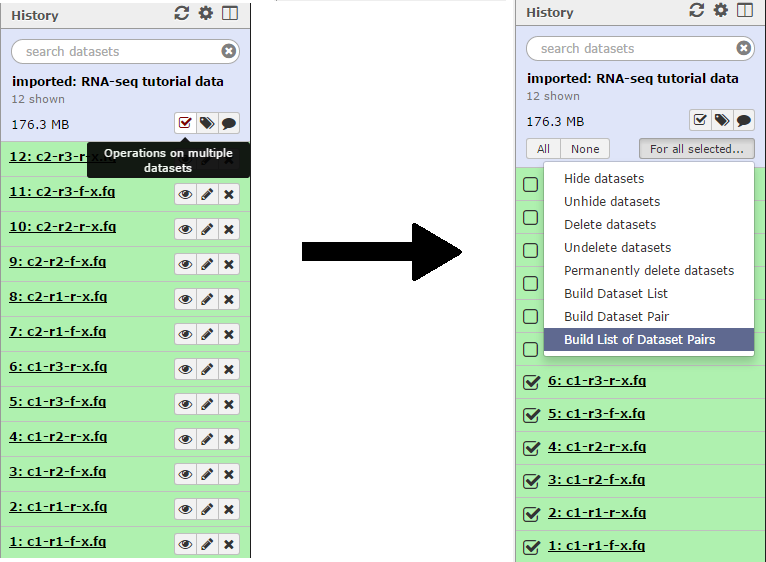
In the unpaired forward box, type “-f-x” and in the unpaired reverse box, type “-r-x”. Then click the Pair these datasets button for the three c1 reads, and to create the list click Create List.

Repeat the previous step for the three condition 2 reads. There should now be a C1 collection and C2 collection data set in the history. Collection 13 will correspond to condition 1, and Collection 14 will correspond to condition 2.
Getting gene annotations (in GTF format) from UCSC
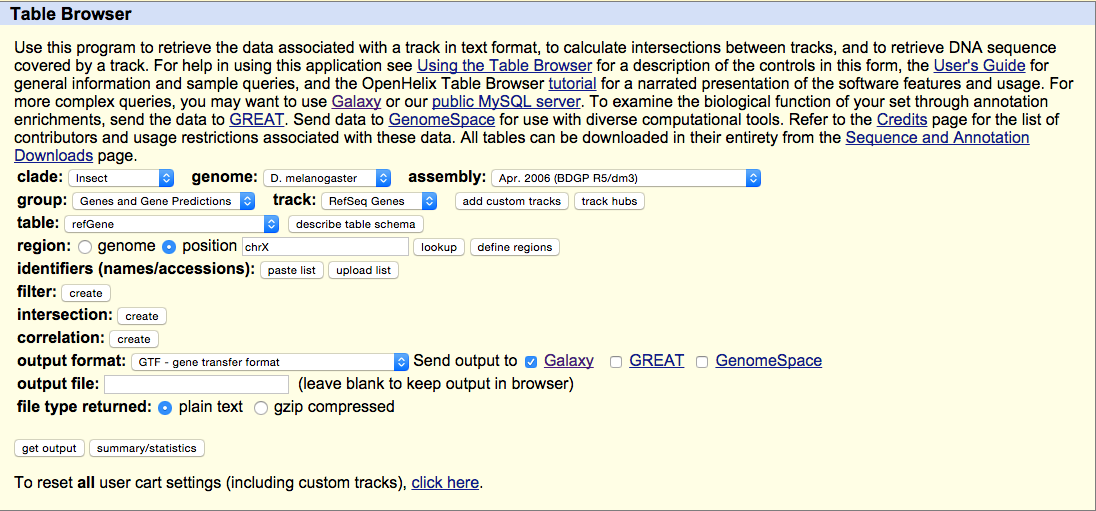
Navigate to the Get Data link in the Tools window and open the UCSC Main table browser.
We are interested in the D. melanogaster Apr. 2006 BDGP R5/dm3 assembly. Change the clade to Insect, genome to D. melanogaster, and assembly to Apr. 2006 BDGP R5/dm3. Set the region to position: chrX. Finally, change the output format to GTF, and click get output then Send query to Galaxy. The GTF annotation file should appear in the history.
Tophat2: Mapping the reads against a reference genome
Navigate to the NGS: RNA Analysis link in the Tools window and open Tophat.
Tophat is used to map the reads in each collection against the dm3 version of Drosophila melanogaster genome. We will first run Tophat on the C1 collection. Set the data type to Paired-end (as collection). Change the mate-pair Mean Inner Distance to 28 and select the dm3 reference genome.
Then, in the Tophat settings to use field, select Full parameter list. Set the Library Type to FR First Strand. Then set Do you want to supply your own junction data –> Yes –> Use Gene Annotation model –> Yes and Galaxy will automatically fill the next field with the annotation file downloaded in the previous step. Then click Execute!
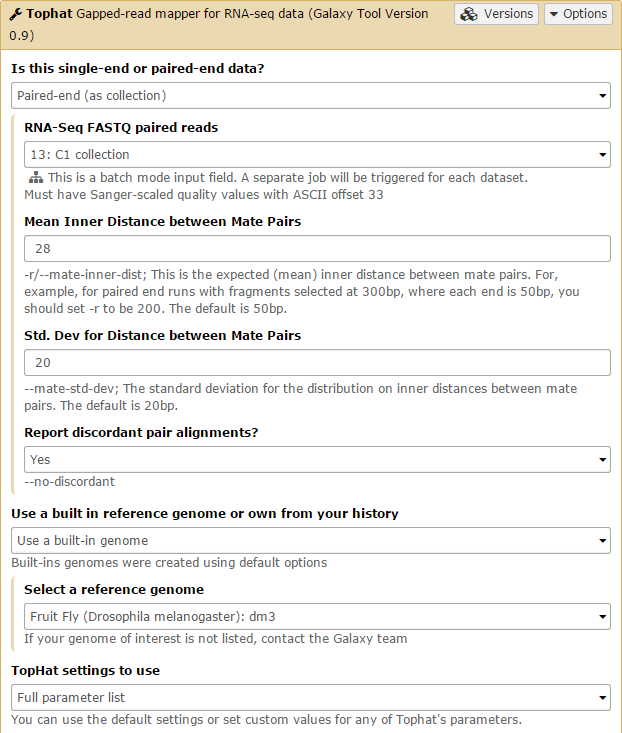
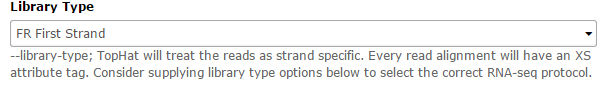
Submit the job again for the C2 collection.
Reconstructing Transcripts with Cufflinks
Navigate to the NGS: RNA Analysis link in the Tools window and open Cufflinks. Click the Dataset collection button to change the input to a dataset collection.
Reads mapped by Tophat are then used as input to Cufflinks – a tool that performs transcript reconstruction and quantification. This is done individually for every replicate (although because our data is bundled in collections this is a painless exercise).
We will first run Cufflinks on the Tophat condition 1 results. The SAM file of aligned RNA-Seq reads input option takes the Tophat accepted_hits output files as input. Set this to the condition 1 accepted_hits (collection 13). Before running, set the Use Reference Annotation field to yes, and Galaxy will automatically fill the field with the annotation file. Also set Perform Bias Correction –> Yes and the dm3 reference genome will automatically be selected.
Then Set advanced Cufflinks options –> Yes. Set Library prep used for input reads to fr-firststrand and change the Inner mean distance to 28, then Execute!
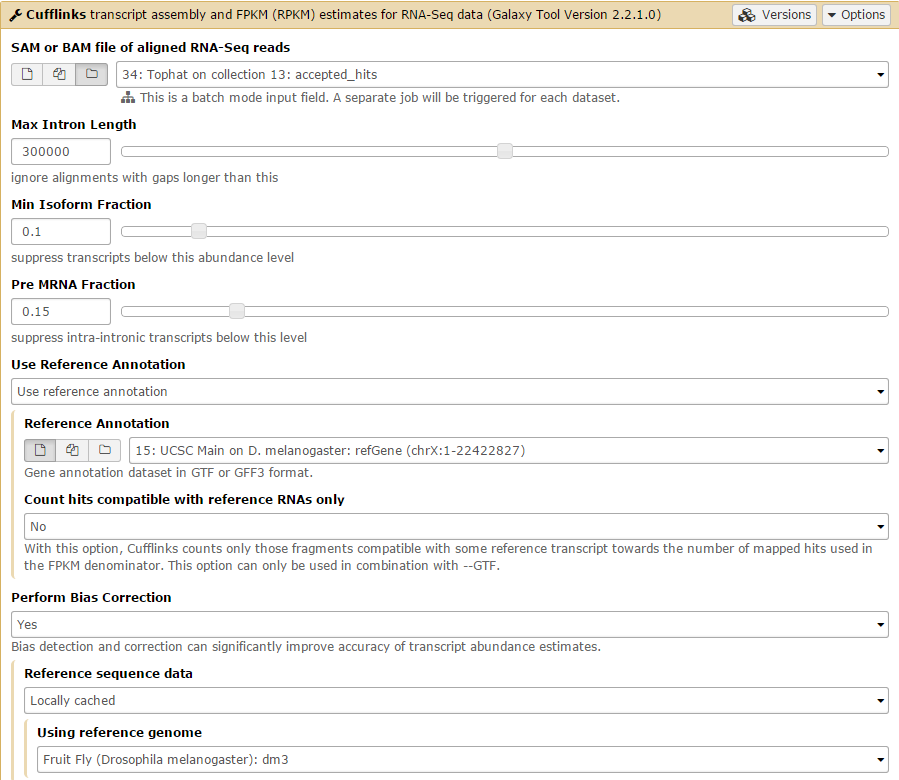

Submit the job again for the C2 collection.
Cuffmerge
Navigate to the NGS: RNA Analysis link in the Tools window and open Cuffmerge. Click the Dataset collection button to change the input to a dataset collection.
We are using Cuffmerge to combine transcript model from all replicates and conditions into a single transcriptome.
There should be 4 datasets available as input in the menu: assembled and skipped transcripts for each condition. Unsurprisingly, we are only interested in the assembled transcripts. Set GTF file(s) produced by Cufflinks to one of the assembled transcripts. Then, add the other assembled transcripts file using the +Insert Additional GTF Inputs (Lists) button. Finally, set the Use Reference Annotation field to yes and click Execute!
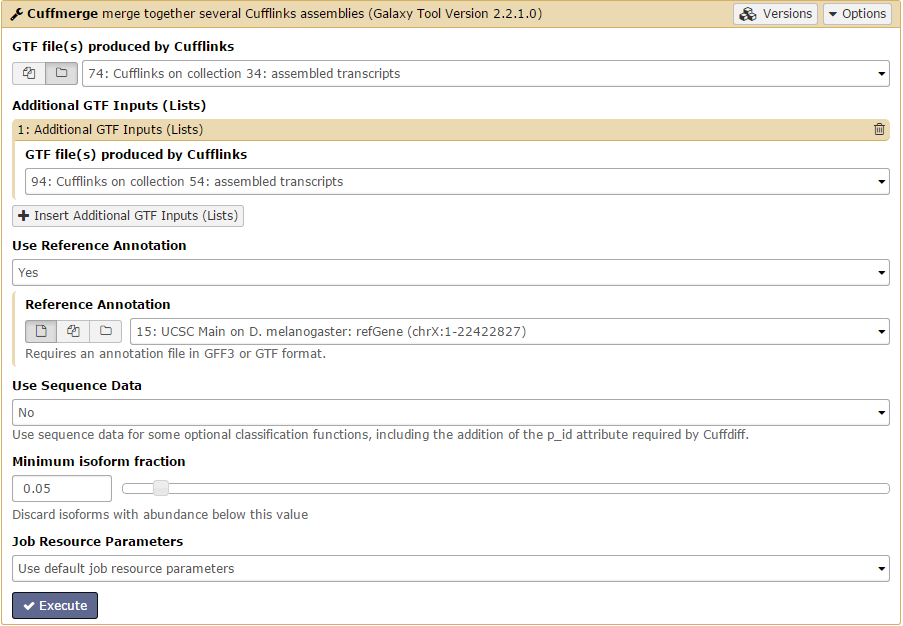
Cuffdiff
Navigate to the NGS: RNA Analysis link in the Tools window and open Cuffdiff.
Finally, we perform differential expression analysis with Cuffdiff using the combined transcriptome and read mapping data.
Cuffdiff will automatically select the Cuffmerge transcript. Set GenerateSQLite to Yes. Then set both conditions: one for C1 on the Tophat collection 13: accepted hits file, and one for C2 on the Tophat collection 14: accepted hits file. Then click Execute.
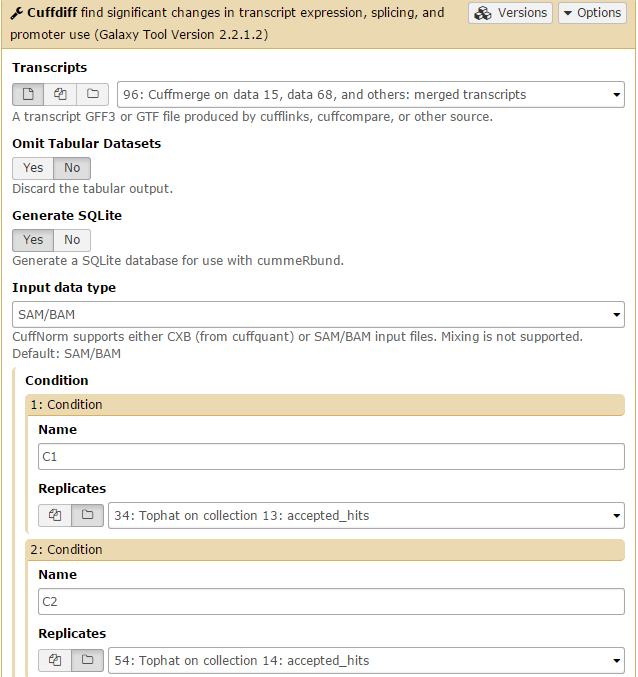
Generating a list of differentially expressed transcripts
Navigate to the Filter and Sort link in the Tools window and open Filter.
Ensure that the Transcript differential expression testing table is selected, and change the filter condition to: c14==’yes’ to remove insignificant results. When the job finishes running, press the Eye button to view the data.
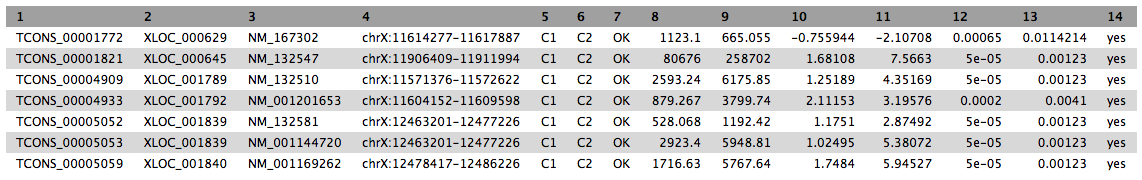
Building a custom track for UCSC genome browser
Navigate to the Build/Graph Data link in the Tools window and open Build a custom track for UCSC genome browser.
The SAM/BAM reads from Tophat can be visualized against the reference genome using this program. Click +Insert Track then click the Dataset collection button to change the input to a dataset collection. Select the condition 1 insertions collection and if desired give your track a custom name. Then click Execute!
To view the track, show hidden datasets then click on one of the custom track files to expand it. Click main in display at UCSC main.