This section explains some of the commonly used file formats in bioinformatics. The information provided here is basic and designed to help users to distinguish the difference between different formats. Please refer user manual or other information resources on web for more details.
FASTA
File format : FASTA
File extensions : file.fa, file.fasta, file.fsa
Example :
>XR_002086427.1 Candida albicans SC5314 uncharacterized ncRNA (SCR1), ncRNA TGGCTGTGATGGCTTTTAGCGGAAGCGCGCTGTTCGCGTACCTGCTGTTTGTTGAAAATTTAAGAGCAAAGTGTCCGGCTCGATCCCTGCGAATTGAATTCTGAACGCTAGAGTAATCAGTGTCTTTCAAGTTCTGGTAATGTTTAGCATAACCACTGGAGGGAAGCAATTCAGCACAGTAATGCTAATCGTGGTGGAGGCGAATCCGGATGGCACCTTGTTTGTTGATAAATAGTGCGGTATCTAGTGTTGCAACTCTATTTTT
Fasta format is a simple way of representing nucleotide or amino acid sequences of nucleic acids and proteins. This is a very basic format with two minimum lines. First line referred as comment line starts with ‘>’ and gives basic information about sequence. There is no set format for comment line. Any other line that starts with ‘;’ will be ignored. Lines with ‘;’ are not a common feature of fasta files. After comment line, sequence of nucleic acid or protein is included in standard one letter code. Any tabulators, spaces, asterisks etc in sequence will be ignored.
FASTQ
File format : FASTQ
File extensions : file.fastq, file.sanfastq, file.fq
Example :
@K00188:208:HFLNGBBXX:3:1101:1428:1508 2:N:0:CTTGTA ATAATAGGATCCCTTTTCCTGGAGCTGCCTTTAGGTAATGTAGTATCTNATNGACTGNCNCCANANGGCTAAAGT + AAAFFJJJJJJJJJJJJJJJJJFJJFJJJJJFJJJJJJJJJJJJJJJJ#FJ#JJJJF#F#FJJ#F#JJJFJJJJJ
Fastq format was developed by Sanger institute in order to group together sequence and its quality scores (Q: phred quality score). In fastq files each entry is associated with 4 lines.
- Line 1 begins with a ‘@‘ character and is a sequence identifier and an optional description.
- Line 2 Sequence in standard one letter code.
- Line 3 begins with a ‘+‘ character and is optionally followed by the same sequence identifier (and any additional description) again.
- Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
Extended description on the fastq format :
Line 1: @K00188:208:HFLNGBBXX:3:1101:1428:1508 2:N:0:CTTGTA

Line 2: ATAATAGGATCCCTTTTCCTGGAGCTGCCTTTAGGTAATGTAGTATCTNATNGACTGNCNCCANANGGCTAAAGT
DNA Sequence
Line 3: +
No comment added
Line 4: AAAFFJJJJJJJJJJJJJJJJJFJJFJJJJJFJJJJJJJJJJJJJJJJ#FJ#JJJJF#F#FJJ#F#JJJFJJJJJ
A quality score (PHRED scale) for each base pair. It indicates how confident we can be that the base was sequenced and identified correctly.
Q = -10log10(p)
where p is the probability that the corresponding base call is incorrect.
| Phred quality score | Probability that the base is called wrong | Accuracy of the base call |
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.90% |
| 40 | 1 in 10000 | 99.99% |
| 50 | 1 in 100000 | 100.00% |
Fastq-sanger holds PHRED score from 0-93 whereas fastq-Illumina provides PHRED scores from 0-62. Rather than giving numeric values of PHRED score they are provided in ASCII character codes from 33 to 126. Why 33 to 126? Because 33 to 126 codes for single characters, so the score can be represented by a single character. Refer the table below.
| Dec | Char | PHRED | Dec | Char | PHRED | Dec | Char | PHRED | Dec | Char | PHRED | Dec | Char | PHRED |
| 33 | ! | 0 | 53 | 5 | 20 | 73 | I | 40 | 93 | ] | 60 | 113 | q | 80 |
| 34 | “ | 1 | 54 | 6 | 21 | 74 | J | 41 | 94 | ^ | 61 | 114 | r | 81 |
| 35 | # | 2 | 55 | 7 | 22 | 75 | K | 42 | 95 | _ | 62 | 115 | s | 82 |
| 36 | $ | 3 | 56 | 8 | 23 | 76 | L | 43 | 96 | ` | 63 | 116 | t | 83 |
| 37 | % | 4 | 57 | 9 | 24 | 77 | M | 44 | 97 | a | 64 | 117 | u | 84 |
| 38 | & | 5 | 58 | : | 25 | 78 | N | 45 | 98 | b | 65 | 118 | v | 85 |
| 39 | ‘ | 6 | 59 | ; | 26 | 79 | O | 46 | 99 | c | 66 | 119 | w | 86 |
| 40 | ( | 7 | 60 | < | 27 | 80 | P | 47 | 100 | d | 67 | 120 | x | 87 |
| 41 | ) | 8 | 61 | = | 28 | 81 | Q | 48 | 101 | e | 68 | 121 | y | 88 |
| 42 | * | 9 | 62 | > | 29 | 82 | R | 49 | 102 | f | 69 | 122 | z | 89 |
| 43 | + | 10 | 63 | ? | 30 | 83 | S | 50 | 103 | g | 70 | 123 | { | 90 |
| 44 | , | 11 | 64 | @ | 31 | 84 | T | 51 | 104 | h | 71 | 124 | | | 91 |
| 45 | – | 12 | 65 | A | 32 | 85 | U | 52 | 105 | i | 72 | 125 | } | 92 |
| 46 | . | 13 | 66 | B | 33 | 86 | V | 53 | 106 | j | 73 | 126 | ~ | 93 |
| 47 | / | 14 | 67 | C | 34 | 87 | W | 54 | 107 | k | 74 | |||
| 48 | 0 | 15 | 68 | D | 35 | 88 | X | 55 | 108 | l | 75 | |||
| 49 | 1 | 16 | 69 | E | 36 | 89 | Y | 56 | 109 | m | 76 | |||
| 50 | 2 | 17 | 70 | F | 37 | 90 | Z | 57 | 110 | n | 77 | |||
| 51 | 3 | 18 | 71 | G | 38 | 91 | [ | 58 | 111 | o | 78 | |||
| 52 | 4 | 19 | 72 | H | 39 | 92 | \ | 59 | 112 | p | 79 |
Based on the base character ( character that represents zero PHRED score ), PHRED scale is often referred as FHRED+33 (ASCII character !) or FHRED+64 (ASCII Character ?). The figure below illustrates the PHRED usage across different sequencing notations.
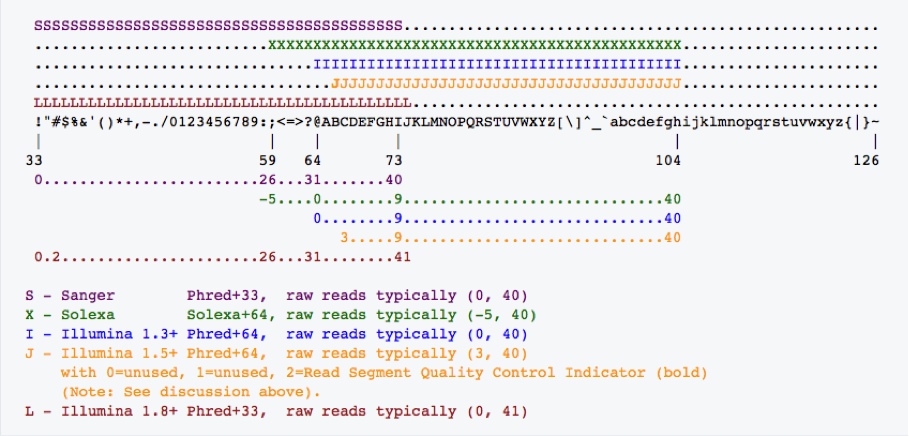
It is critical to figure out the PHRED score type used in .fastq files before processing them. Current Illumina .fastq files have +33 PHRED score. Please refer https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2847217/ for more details.
SAM (Sequence Alignment Map)
File format : SAM
File extensions : file.sam
Example :
1:497:R:-272+13M17D24M 113 1 497 37 37M 15 100338662 0 CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 19:20389:F:275+18M2D19M 99 1 17644 0 37M = 17919 314 TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9 RG:Z:UM0098:1 XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 19:20389:F:275+18M2D19M 147 1 17919 0 18M2D19M = 17644 -314 GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:18^CA19 9:21597+10M2I25M:R:-209 83 1 21678 0 8M2I27M = 21469 -244 CACCACATCACATATACCAAGCCTGGCTGTGTCTTCT <;9<<5><<<<><<<>><<><>><9>><>>>9>>><> XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:5 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:35
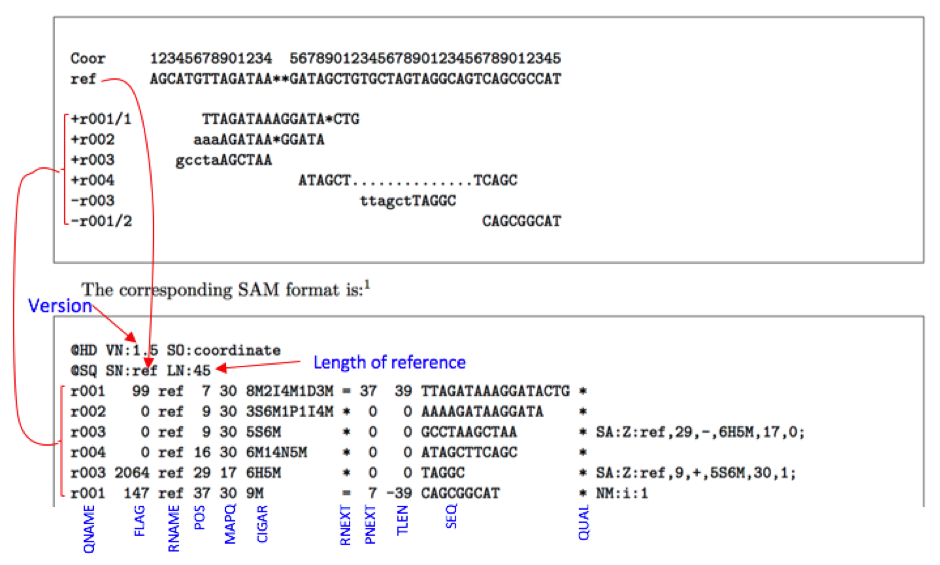
The SAM Format is a text format for storing sequence data in a series of tab delimited ASCII columns. Most often it is generated as a human readable version of its sister BAM format, which stores the same data in a compressed, indexed, binary form.
SAM format files are generated following mapping of the reads to reference sequence. It is TAB-delimited text format with header and a body. Header lines start with ‘@’ while alignment lines do not. Header hold generic information on SAM file along with version information, if the file is sorted, information on reference sequence, etc. The alignment records constitute the body of the file. Each alignment line/record has 11 mandatory fields describing essential alignment information.
Some terminology used in SAM manual:
Template: The DNA fragment that was measured
Reads: Depending on the methodology a template may produce one or more reads. These reads may cover the entire template or just a subsection of it. Reads originating from the same template typically cover different parts of the template, and, may represent the template itself or the reverse complement of it.
Segments: Each read may produce one or more alignments that in turn will have aligned regions called segments. From these segments it may be possible the infer the size of the original template.
source: http://samtools.github.io/hts-specs/SAMv1.pdf
Fields:
Col. 1 QNAME:
Query NAME. Reads/segments having identical QNAME are regarded to come from the same template. A QNAME ‘*’ indicates the information is unavailable. In a SAM file, a read may occupy multiple alignment lines, when its alignment is chimeric .
Col. 2 FLAG:
Combination of bitwise flags.
| BIT | Description | |
| 1 | 0x1 | template having multiple segments in sequencing |
| 2 | 0x2 | each segment properly aligned according to the aligner |
| 4 | 0x4 | segment unmapped |
| 8 | 0x8 | next segment in the template unmapped |
| 16 | 0x10 | SEQ being reverse complemented |
| 32 | 0x20 | SEQ of the next segment in the template being reverse complemented the first segment in the template |
| 64 | 0x40 | the first segment in the template |
| 128 | 0x80 | the last segment in the template |
| 256 | 0x100 | secondary alignment |
| 512 | 0x200 | not passing filters, such as platform/vendor quality controls |
| 1024 | 0x400 | PCR or optical duplicate |
| 2048 | 0x800 | supplementary alignment |
Col. 3 RNAME:
Name of reference sequence. It generally refers to chromosome number.
Col. 4 POS:
Leftmost mapping position of the first matching base in read. It has 1-based indexing. If pos is set as 0, it represents a unmapped read. For a read pair READ1/1 and READ1/2 and single Read2
Reference: GTACGACTGACTAGACGATAC**GTAACGATCAGTCTCGATAGCGATAGGCTAGCTAGCAGCTAGCAG 12345678901234567890123456789012345678901234567890123456789012345678 Read1/1 ACGACTGA -Read1/2 CGATAGC Read2 ccccGATACTAGTAA*GAT..GTCT Pos values 3 38
Col. 5 MAPQ:
It indicates MAPpping Quality. MAPQ= -10log10(Probability of mapping position being wrong). MAPQ=255 indicates mapping quality is unavailable.
Col. 6 CIGAR:
A string that describes alignment.
| OP | BAM | Description |
| M | 0 | alignment match (can be a sequence match or mismatch) |
| I | 1 | insertion to the reference |
| D | 2 | deletion from the reference |
| N | 3 | skipped region from the reference |
| S | 4 | soft clipping (clipped sequences present in SEQ) |
| H | 5 | hard clipping (clipped sequences NOT present in SEQ) |
| P | 6 | padding (silent deletion from padded reference |
| = | 7 | sequence match |
| X | 8 | sequence mismatch |
Difference between H and S is that if the mismatch sequence is reported as part of read sequence in alignment file it is a soft clipping. Often mismatch region matches somewhere else in reference sequence and in that case the mismatch region is removed from reported read sequence in alignment and is referred as Hard clipping.
Reference: GTACGACTGACTAGACGATAC**GTAACGATCAGTCTCGATAGCGATAGGCTAGCTAGCAGCTAGCAG 12345678901234567890123456789012345678901234567890123456789012345678 Read2 ccccGATACTAGTAA*GAT..GTCT
CIGAR value of Read2 in POS example will be:
cccc does not match anywhere else in reference so soft clipping. , 5 ( GATAC) Matches, 2 (TA) Insertion, 4(GTAA) Matches, 1(*) Deletion, 3 (GAT) Matches, 2(..)Skipped region from reference(N), 4(GTCT) Matches
CIGAR Strings:
Read2 : 5S2M2I4M1D3M2N4M
Col. 7 RNEXT , Col. 8 PNEXT:
RNEXT and PNEXT is to know the reference and position of a paired end read’s partner for visualisation tools. RNEXT is the name of the chromosome or contig to which the next template in a pair aligns. RNEXT of value ‘=‘ means align to same reference and ‘*’ represent no information available (single end sequencing). PNEXT where the other read of the pair aligns (Information unavailable =0, Otherwise POS value of pair).
Reference: GTACGACTGACTAGACGATAC**GTAACGATCAGTCTCGATAGCGATAGGCTAGCTAGCAGCTAGCAG 12345678901234567890123456789012345678901234567890123456789012345678 Read1/1 ACGACTGA -Read1/2 CGATAGC Read2 ccccGATACTAGTAA*GAT..GTCT
Read1/1 and Read1/2 are pair and Read 3 is unpaired. So the RNEX and PNEXT values will be
READ RNEXT PNEXT TLEN
Read1/1 = 38 42
Read1/2 = 3 -42
Read3 * 0 0
Col. 9 TLEN : Observed Template LENgth
It represents the length of reference that is covered by pair end reads. The distance between leftmost mapped base to rightmost mapped base in paired reads. For unpaired reads it is 0.
Col. 10 SEQ: Sequence of the read or Segment.
Col. 11 QUAL: PHRED score values of read. If ‘*’ no values are stored.
Example:
BAM
File format : BAM
File extensions : file.bam
A BAM (Binary Alignment/Map) file is the compressed binary version of the Sequence Alignment/Map (SAM), a compact and indexable representation of nucleotide sequence alignments. The data between SAM and BAM is exactly same. Being Binary BAM files are small in size and ideal to store alignment files. Require samtools to view the file.
VCF (Variant Calling Format/File)
File format : VCF
File extensions : file.vcf
Example :
##fileformat=VCFv4.2 ##fileDate=20090805 ##source=myImputationProgramV3.1 ##reference=file:///seq/references/1000GenomesPilot-NCBI36.fasta ##contig=<ID=20,length=62435964,assembly=B36,md5=f126cdf8a6e0c7f379d618ff66beb2da,species="Homo sapiens",taxonomy=x> ##phasing=partial ##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data"> ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth"> ##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency"> ##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele"> ##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP membership, build 129"> ##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership"> ##FILTER=<ID=q10,Description="Quality below 10"> ##FILTER=<ID=s50,Description="Less than 50% of samples have data"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002 NA00003 20 14370 rs6054257 G A 29 PASS NS=3;DP=14;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51 1/1:43:5:.,. 20 17330 . T A 3 q10 NS=3;DP=11;AF=0.017 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3 0/0:41:3 20 1110696 rs6040355 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 2/2:35:4 20 1230237 . T . 47 PASS NS=3;DP=13;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:51,51 0/0:61:2 20 1234567 microsat1 GTC G,GTCT 50 PASS NS=3;DP=9;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2 1/1:40:3
VCF is a text file format with a header (information VCF version, sample etc) and data lines constitute the body of file.
HEADER:
This contains meta-information and is included after ‘##’ string. It is recommended to include INFO, FILTER and FORMAT entries for a better explanation of the data field.
Metadata format:
##INFO=<ID=ID,Number=number,Type=type,Description="description",Source="source",Version="version"> ##FILTER=<ID=ID,Description="description">##FORMAT=<ID=ID,Number=number,Type=type,Description="description">
Other information like alternate allele, assembly field, Contig field, sample field, pedigree field can also be included.
DATA FIELD:
Data lines have 8 mandatory columns.
#CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO.
VCF format has very well explained manual available at https://samtools.github.io/hts-specs/VCFv4.2.pdf .
GFF (General Feature Format or Gene Finding Format)
File format : GFF
File extensions : file.gff2, file. gff3, file.gff
Example :
GFF2:
browser position chr22:10000000-10025000 browser hide all track name=regulatory description="TeleGene(tm) Regulatory Regions" visibility=2 chr22 TeleGene enhancer 10000000 10001000 500 + . touch1 chr22 TeleGene promoter 10010000 10010100 900 + . touch1 chr22 TeleGene promoter 10020000 10025000 800 - . touch2
GFF3:
It has first 8 fields like GFF2 but differs in field 9 in assigning attributes. 2 are highlighted here.
(a) GFF3 has better nesting feature. Links features to parent tag
##gff-version 3 ctg123 . mRNA 1300 9000 . + . ID=mrna0001;Name=sonichedgehog ctg123 . exon 1300 1500 . + . ID=exon00001;Parent=mrna0001 ctg123 . exon 1050 1500 . + . ID=exon00002;Parent=mrna0001 ctg123 . exon 3000 3902 . + . ID=exon00003;Parent=mrna0001 ctg123 . exon 5000 5500 . + . ID=exon00004;Parent=mrna0001 ctg123 . exon 7000 9000 . + . ID=exon00005;Parent=mrna0001
(b) The most general way of representing a protein-coding gene is the so-called “three-level gene.” The top level is a feature of type “gene” which bundles up the gene’s transcripts and regulatory elements. Beneath this level are one or more transcripts of type “mRNA”. This level can also accommodate promoters and other cis-regulatory elements. At the third level are the components of the mRNA transcripts, most commonly CDS coding segments and UTRs. This example shows how to represent a gene named “EDEN” which has three alternatively-spliced mRNA transcripts:
ctg123 example gene 1050 9000 . + . ID=EDEN;Name=EDEN;Note=protein kinase ctg123 example mRNA 1050 9000 . + . ID=EDEN.1;Parent=EDEN;Name=EDEN.1;Index=1 ctg123 example five_prime_UTR 1050 1200 . + . Parent=EDEN.1 ctg123 example CDS 1201 1500 . + 0 Parent=EDEN.1 ctg123 example CDS 3000 3902 . + 0 Parent=EDEN.1 ctg123 example CDS 5000 5500 . + 0 Parent=EDEN.1 ctg123 example CDS 7000 7608 . + 0 Parent=EDEN.1 ctg123 example three_prime_UTR 7609 9000 . + . Parent=EDEN.1 ctg123 example mRNA 1050 9000 . + . ID=EDEN.2;Parent=EDEN;Name=EDEN.2;Index=1 ctg123 example five_prime_UTR 1050 1200 . + . Parent=EDEN.2 ctg123 example CDS 1201 1500 . + 0 Parent=EDEN.2 ctg123 example CDS 5000 5500 . + 0 Parent=EDEN.2 ctg123 example CDS 7000 7608 . + 0 Parent=EDEN.2 ctg123 example three_prime_UTR 7609 9000 . + . Parent=EDEN.2 ctg123 example mRNA 1300 9000 . + . ID=EDEN.3;Parent=EDEN;Name=EDEN.3;Index=1 ctg123 example five_prime_UTR 1300 1500 . + . Parent=EDEN.3 ctg123 example five_prime_UTR 3000 3300 . + . Parent=EDEN.3 ctg123 example CDS 3301 3902 . + 0 Parent=EDEN.3 ctg123 example CDS 5000 5500 . + 1 Parent=EDEN.3 ctg123 example CDS 7000 7600 . + 1 Parent=EDEN.3 ctg123 example three_prime_UTR 7601 9000 . + . Parent=EDEN.3
Source: http://gmod.org/wiki/GFF3
GFF (General Feature Format or Gene Finding Format). GFF can be used for any kind of feature (Transcripts, exon, intron, promoter, 3’ UTR, repeatitive elements etc) associated with the sequence, whereas GTF is primarily for genes/transcripts. GFF3 is the latest version and an improvement over GFF2 format. However, many databases are still not equipped to handle GFF3 version. The differences will be explained later in text.
The GFF format has 9 mandatory columns and they are TAB separated. The 9 columns are as follows.
Col. 1 Reference Sequence:
This is the ID of reference sequence used to establish coordinate system for annotation. Usually chromosome name or number.
Col. 2 Source:
This explains how the feature annotation is derived. The source is a free text qualifier intended to describe the algorithm or operating procedure that generated this feature. Typically this is the name of a piece of software, such as “Genescan” or a database name, such as “Genbank.” In effect, the source is used to extend the feature ontology by adding a qualifier to the type creating a new composite type that is a subclass of the type in the type column. It is not necessary to specify a source. If there is no source, put a “.” (a period) in this field.
Col. 3 Feature:
The feature type name, like “gene” or “exon”. In a well-structured GFF file, all the children(exons, introns etc) features always follow their parents(Transcript) feature line. This way they are part of a single block
Col. 4 Start:
Genomic Start of the feature.
Col. 5 End:
Genomic Start of the feature
Col. 6 Score:
Numeric value that generally indicates the confidence of the source on the annotated feature. A value of “.” (a dot) is used to define a null value. The semantics of the score are ill-defined. In GFF3 format,It is strongly recommended that E-values be used for sequence similarity features, and that P-values be used for ab initio gene prediction features. If there is no score, put a “.” (a period) in this field.
Col. 7 Strand:
Field that indicates the sense strand of the feature. “+’ :Watson strand and ‘-‘: crick strand. ‘?’ can be used for features whose strandedness is relevant, but unknown.
Col. 8 Frame (GFF2 and GTF) or Phase (GFF3):
For features of type “CDS”, the phase indicates where the feature begins with reference to the reading frame. The phase is one of the integers 0, 1, or 2, indicating the number of bases that should be removed from the beginning of this feature to reach the first base of the next codon. In other words, a phase of “0” indicates that the next codon begins at the first base of the region described by the current line, a phase of “1” indicates that the next codon begins at the second base of this region, and a phase of “2” indicates that the codon begins at the third base of this region. This is NOT to be confused with the frame, which is simply start modulo 3. If there is no phase, put a “.” (a period) in this field.
For forward strand features, phase is counted from the start field. For reverse strand features, phase is counted from the end field.
The phase is required for all CDS features.
Explained let say ### and *** represent consecutive exons.
CTG C is first base (0), T is second base (1), G is third base(2)
Previous exon Phase value next Exon 5’ 3’ …###***###***### 0 ***###***###***#… …##***###***###* 1 **###***###***##… …#***###***###** 2 *###***###***###…
Col. 9 Attribute or Group field:
All lines with the same group are linked together into a single item. The group field is a challenge. It is used in several distinct ways:
- to group together a single sequence feature that spans a discontinuous range, such as a gapped alignment.
- to name a feature, allowing it to be retrieved by name.
- to add one or more notes to the annotation.
- to add an alternative name
Problems with GFF2:
One of GFF2’s problems is that it is only able to represent one level of nesting of features. This is mainly a problem when dealing with genes that have multiple alternatively-spliced transcripts. GFF2 is unable to deal with the three-level hierarchy of gene → transcript → exon. Most people get around this by declaring a series of transcripts and giving them similar names to indicate that they come from the same gene.
The second limitation is that while GFF2 allows you to create two-level hierarchies, such as transcript → exon, it doesn’t have any concept of the direction of the hierarchy. So it doesn’t know whether the exon is a subfeature of the transcript, or vice-versa. This means you have to use “aggregators” to sort out the relationships. This is a major pain in the neck. For this reason, GFF2 format has been deprecated in favor of GFF3 format databases.
GTF (Gene Transfer format)
File format : GTF
File extensions : file.gtf
Example :
AB000381 Twinscan exon 150 200 . + . gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan exon 300 401 . + . gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan CDS 380 401 . + 0 gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan exon 501 650 . + . gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan CDS 501 650 . + 2 gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan exon 700 800 . + . gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan CDS 700 707 . + 2 gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan exon 900 1000 . + . gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan start_codon 380 382 . + 0 gene_id "AB000381.000"; transcript_id "AB000381.000.1"; AB000381 Twinscan stop_codon 708 710 . + 0 gene_id "AB000381.000"; transcript_id "AB000381.000.1";
GTF has the same format as GFF files. It has the same 9 fields that describe the gene/ transcript related features. The group/attribute field has been expanded into a list of attributes. Each attribute consists of a type/value pair. Attributes must end in a semi-colon, and be separated from any following attribute by exactly one space. The attribute list must begin with the two mandatory attributes:
gene_id value: A globally unique identifier for the genomic source of the sequence.
transcript_id value: A globally unique identifier for the predicted transcript.
A very good account of GTF file is present at http://mblab.wustl.edu/GTF2.html